
RESEARCH ARTICLE
Single Nucleotide Variants in A Family of Monozygotic Twins Discordant for the Phenotype Congenital Megaureter: A Genomic Analysis
Augusto C. Soares dos Santos Junior1, 2, *, Luciana B. Rodrigues4, Raony G. Corrêa Do Carmo Lisboa Cardenas1, Patricia G.P. Couto1, Luiz A. Cunha de Marco1, Eduardo A. Oliveira3, Debora M. de Miranda1, Ana C. Simoes e Silva3
Article Information

Identifiers and Pagination:
Year: 2017Volume: 10
First Page: 11
Last Page: 19
Publisher ID: TOUNJ-10-11
DOI: 10.2174/1874303X01710010011
Article History:
Received Date: 07/11/2016Revision Received Date: 01/03/2017
Acceptance Date: 11/03/2017
Electronic publication date: 30/05/2017
Collection year: 2017

open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: https://creativecommons.org/licenses/by/4.0/legalcode. This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Introduction:
Congenital megaureter constitutes the second most frequent cause of hydronephrosis in children. There is still much debate on what extent environmental or genetic factors are involved in the pathogenesis of congenital megaureter.
Objectives:
This study aimed at investigating a pair of monozygotic twins discordant for the expression of bilateral congenital megaureter using the whole exome sequencing technique.
Methods:
Peripheral blood DNA was extracted and then sequenced using the whole exome technique from a pair of twins discordant for the presence of bilateral congenital refluxing unobstructed megaureter, his parents and a set of 11 non-related individuals with confirmed diagnosis of congenital megaureter. The DNA of the set of 11 non-related individuals was pooled in three groups. The monozygotic twins and their parents had DNA samples sequenced separately. Sanger validation was performed after data was filtered.
Results:
In the proband were identified 256 candidate genes, including TBX3, GATA6, DHH, LDB3, and HNF1, which are expressed in the urinary tract during the embryonic period. After Sanger validation, the SNVs found in the genes TBX3, GATA6, DHH and LDB3 were not confirmed in the proband. The SNV chr17:36104650 in the HNF1b gene was confirmed in the proband, his twin brother and the mother, however was not found in the pool of 11 non-related individuals with congenital megaureter.
Conclusion:
Due to the possible complex causative network of genetic variations and the challenges regarding the use of the whole exome sequencing technique we could not unequivocally associate the genetic variations identified in this study with the development of the congenital megaureter.
1. INTRODUCTION
Congenital anomalies of the kidney and urinary tract [CAKUT] represent a broad range of disorders that result from abnormalities of the urinary collecting system, abnormal embryonic migration of the kidneys or abnormal renal parenchyma development [1, 2]. Megaureter constitutes one of the phenotypes of CAKUT and represents a condition whereby the diameter of the ureter is abnormally superior to 7 mm [3-5]. Congenital megaureter constitutes the second most frequent cause of hydronephrosis in children with an estimated incidence of 0.36 cases for every 1,000 newborns. Conceptually it can be classified according to the presence or the absence of vesicoureteral reflux and obstruction [6].
There is still much debate on what extent environmental, epigenetic or genetic factors are implicated in the pathogenesis of CAKUT. The current most accepted theory supports the influence of a complex interaction between environmental, epigenetic and genetic factors, suggesting that the pathogenesis of CAKUT is multifactorial [7]. However, uncertainty is especially true when the debate comes specifically to the factors involved in the development of congenital megaureter. Some authors have hypothesized that variations in specific regions of the genome could imbalance the mechanisms related to the expression of cytokines that modulate the ureter morphogenesis [8-10]. In other words, changes affecting single nucleotides could be a possible mechanism to trigger abnormal pathways, which would eventually signal to the development of the megaureter [11-13]. In this context, several genes have emerged as potential candidates [14]. Alterations with single nucleotides are detected by whole-exome sequencing [WES], a technique used to determine the DNA sequence for protein coding regions known as exons [14-22]. The exons represents only about 1.5% of the genome, however it is currently believed that 85% of all genetic diseases derive from modifications in this region of the genome [23, 24].
Taking all these factors together, the study of discordant monozygotic twins has become of special interest in the study of patients with congenital megaureter, since this approach offers the opportunity to control for potential confounders, including differences in genetic background or in utero environment [25-28]. Therefore, this study aimed to investigate a pair of monozygotic twins discordant for the expression of bilateral congenital megaureter and for the presence of genetic variants by using the WES technique. Validation with an external sample of non-related cases of megaureter was also used to further assess the possible association between gene and disease.
2. PATIENTS AND METHODS
2.1. Patients and Clinical Assessment
This study included twins discordant for the presence of bilateral congenital refluxing unobstructed megaureter, his parents and a set of 11 non-related individuals with confirmed diagnosis of congenital megaureter (see Fig. (1) and Table 1). Ultrasound [US] scan was performed in all cases after 28 weeks of gestation. Megaureter was considered to be present in case of an ureter diameter superior to 7 mm in fetal US [3, 5]. Patients were followed up at the Pediatric Nephrology Unit of the Hospital das Clínicas - Universidade Federal de Minas Gerais [UFMG], Brazil and all of them underwent postnatal image and laboratory investigation according to a systematic protocol as detailed described elsewhere [29].
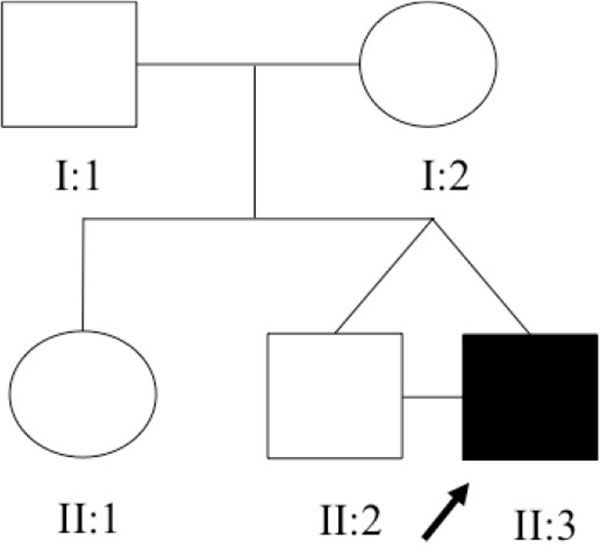 |
Fig. (1). Pedigree of the family under study. |
| Date of birth | Gender | Phenotype | |
|---|---|---|---|
| 1. | 09/06/2001 | Male | Congenital right nonrefluxing unobstructed megaureter |
| 2. | 22/05/2002 | Female | Congenital right nonrefluxing unobstructed megaureter |
| 3. | 06/04/1992 | Male | Congenital right nonrefluxing unobstructed megaureter |
| 4. | 02/04/2005 | Male | Congenital left nonrefluxing unobstructed megaureter |
| 5. | 21/11/2006 | Male | Congenital left nonrefluxing unobstructed megaureter |
| 6. | 14/04/1989 | Female | Congenital bilateral nonrefluxing unobstructed megaureter |
| 7. | 01/12/2007 | Female | Congenital bilateral nonrefluxing unobstructed megaureter |
| 8. | 16/05/1999 | Female | Congenital left nonrefluxing unobstructed megaureter |
| 9. | 14/03/1999 | Female | Congenital bilateral nonrefluxing unobstructed megaureter |
| 10. | 15/04/2004 | Male | Congenital left nonrefluxing unobstructed megaureter |
| 11. | 17/10/2007 | Male | Congenital left nonrefluxing unobstructed megaureter |
The local Ethics Committee approved this study in accordance with the principles outlined in the Declaration of Helsinki. Informed consent was obtained from the parents or responsible for all participants. The study protocol did not interfere with any medical recommendation or prescription.
The family under study was constituted of 3 siblings (Fig. 1). The older sister refused to have blood samples collected and consequently she was not included in the genomic analysis. All family members, except the proband, had normal renal function and normal kidney and urinary tract anatomy. The twins, born in 22/08/2008 at 38 ± 1 weeks of gestation, were monozygotic, monochorionic and diamniotic. The first signs of CAKUT in the proband were seen during the US screening at 29 ± 1 weeks of gestation when his fetal biometry revealed left kidney ectasy and an increase bladder volume. In a spite of these prenatal US findings, the proband was not primarily referred for further investigations or treatment. A retrospective evaluation of pregnancy history did not identify exposure to known environmental risk factors for fetal anomalies. During his first month of life, in 19/09/2008, the proband developed urinary tract infection followed by sepsis and acute renal failure. He was then hospitalized in an intensive care unit and started peritoneal dialysis. Interruption of the dialysis sessions occurred after infection control and partial recovery of the renal function. During the hospital stay, a miccional urethrocystography revealed bilateral megaureter associated with grade V vesicoureteral reflux, as shown in (Fig. 2). In 13/10/2008, the proband underwent bilateral surgical re-implant of the ureters and was then referred to outpatient care at the Pediatric Nephrology Unit. At the time of recruitment, the two monozygotic twins were five years old and the proband exhibited CKD stage III.
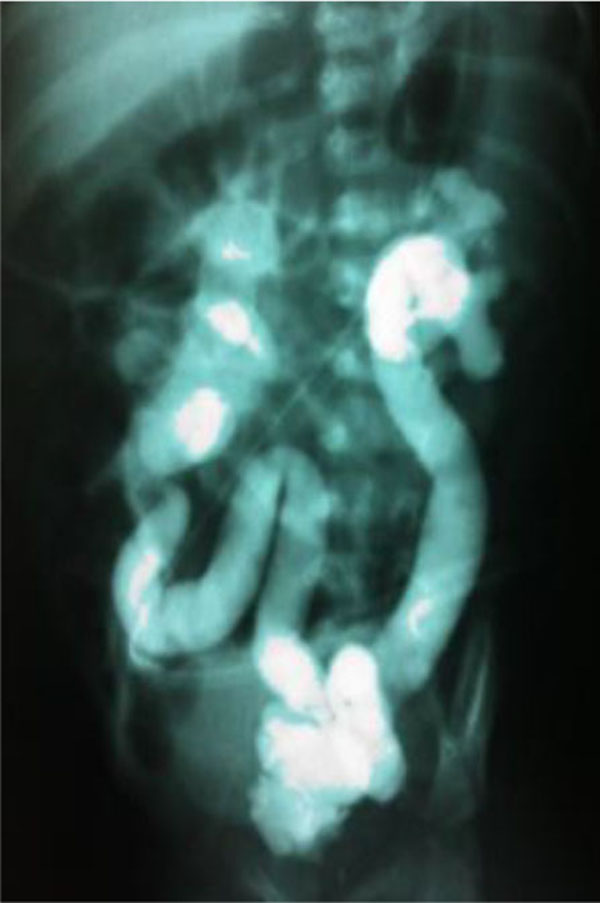 |
Fig. (2). Phenotype of the proband [miccional urethrocystography]. |
2.2. DNA Extraction and Whole Exome Sequencing
After the signature of the informed consent, all participants underwent an intravenous puncture to collect 15 ml of peripheral blood samples. DNA was extracted from peripheral blood lymphocytes by the method described by Lahiri and Nurnberger [30]. All samples were checked for quality control and purity using a Nanodrop spectrophotometer [Thermo Scientific, Waltham, MA]. DNA samples were stored at -20°C until usage.
Peripheral blood DNA was extracted from the subjects under study and underwent whole exome capturing and sequencing using the Roche NimbleGen V2 chip [Madison, Wisconsin] or Nextera [San Diego, California] with the Illumina HiSeq2000 sequencing platform [San Diego, California]. The monozygotic twins and their parents had DNA samples sequenced separately. The DNA of the set of 11 non-related individuals with confirmed diagnosis of congenital megaureter was pooled in three groups. Array capture was used to isolate relevant human genes [Roche/NimblegenSeqCap EZ Human Exome Library v2.0]. These genes were sequenced on the Illumina HiSeq2000 platform [19].
2.3. Variantion Calling and Annotation
Raw sequence files were prepared using the Genome Analysis Tool Kit [GATK] for each of the sequenced samples. Each fastq file was aligned against the human hg19/GRCh37 reference genome. PCR duplicates were removed using Picard [http://picard.sourceforge.net/], reads around known and detected indels were realigned, and base quality was recalibrated using GATK. In order to call variants from the processed BAM files, a variant calling pipeline from GATK was applied. All generated VCF files were analyzed using Mendel MD software developed by the Clinical Genomic Laboratory of UFMG and available at http://mendel.medicina.ufmg.br [31].
2.4. Filtering Data
Previously described single nucleotide variants [SNVs], by November 2015, in the following databases were excluded: Project 1000 genomes [32], Exome Variant Server NHLBI GO Exome Sequencing Project [ESP] and dbSNP database [33].
To prioritize the high quality variants, SNVs fulfilling the following criteria were excluded: [i] variants within intergenic, intronic, and UTR regions; [ii] synonymous mutations; [iii] variants with phred quality score minor than 20; [iv] variants with conservation score [phyloP] inferior to 3.
Sanger validation was performed after analysis of the literature for the identified genes regarding its known function and expression in kidney related structures during the embrionary period. The final set of selected variants was visually inspected using Integrative Genomics Viewer [IGV version 2.3.40] [34]. The prediction of amino acid substitution on the biological function of the protein [35] was evaluated using the PolyPhen2 [36].
2.4.1. Validation of Data
Sanger sequencing was utilized in the analysis to confirm SNVs in the selected genes. Amplification products of appropriate size were identified using polyacrylamide gel electrophoresis. Products were purified using the QIAquick PCR purification kit [QIAGEN, Milano, Italy] and then submitted to sequencing reaction using both forward and reverse primers with the ABI BigDye Terminator Cycle Sequencing Kit v3.1 on an ABI PRISM 3130 Genetic Analyzer [Applied Biosystems, Foster City, USA]. Each read was aligned to the reference sequence, and mutations were identified with the Sequencer software [GeneCodes, Ann Arbor, MI].
3. RESULTS
In the proband, 70.8 million reads were sequenced with a mean read length of 74 base pairs [bp]. The mapped bp mean error rate was 0.41%, and the resultant mean base coverage was 71.8 times. A total of 95.5% of the sequenced bases were covered at least eight times by band reads. The observed number of SNVs, nonsynonymous mutations, read depth and read quality for each individual in this study and the pools are described in (Table 2).
| SNVs | Non synonymous mutations | Read depth* | Read quality* | |
|---|---|---|---|---|
| Proband | 40,123 | 15,028 | 61.23 [1 / 200] | 963.4 [10.2 / 7,844.7] |
| Proband father | 43,104 | 15,968 | 65.2 [1 / 200] | 1,036.8 [10.2 / 7,990.7] |
| Proband mother | 38,141 | 14,032 | 63.2 [1 / 200] | 1,035.6 [10.2 / 7,836.7] |
| Proband brother | 41,008 | 14,961 | 73.2 [1 / 200] | 1,138.7 [10.2 / 7,679.7] |
|
Megaureter pool 1 Megaureter pool 2 Megaureter pool 3 |
42,983 53,862 54,267 |
15,606 19,662 19,871 |
71.7 [1 / 200] 74.3 [1 / 200] 78.8 [1 / 200] |
1,142.4 [10.2 / 7,838.7] 752.0 [10.2 / 7,959.7] 724.2 [10.2 / 7,642.7] |
The proband had 40,123 SNVs identified. The proband and his twin shared 35,990 [88.96%] SNVs. From these, 35,695 [99.2%] SNVs resulted in common genotypes. Similarly, the proband shared 29,516 [59%] and 27,114 [52.58%] of SNVs and 5,842 [20%] and 6,019 [22%] common genotypes, with his father and mother, respectively.
After data filtering, we found in the proband 256 candidate genes with variants, (Table 3). Among these, we identify genes expressed in urinary tract during embrionary period including TBX3, GATA6, DHH, LDB3, and HNF1b. The variants in these genes were selected for Sanger validation, (Table 4).
| Frequency | |
|---|---|
| Total number of SNVs in the proband | 40,123 |
| Number of previously described SNVs * [excluded] | 36,165 |
| SNVs identified as coding silent, intergenic, intronic, UTR and synonymous mutations, quality score < 20, PhyloP score < 3 [excluded] | 3,702 |
| Remaining candidate SNVs | 256 |
| Gene | Chr | Chr position [hg19] | Códon change | Zygosity | AA change | PolyPhen-2 [36] |
|---|---|---|---|---|---|---|
| TBX3 | 12 | 115110015 | G>T | Heterozygous | S621R | 0,534 |
| DHH | 12 | 49483730 | G>A | Heterozygous | A368V | 0,898 |
| GATA6 | 18 | 19751573 | C>A | Heterozygous | S156R | 0,761 |
| LDB3 | 10 | 88476146 | T>C | Heterozygous | S437P | 0,969 |
| HNF1b | 17 | 36070591 | T>G | Heterozygous | G76C | 0,966 |
The SNVs found in the genes TBX3, GATA6, DHH and LDB3 were not confirmed in the proband. The heterozygote variant in the HNF1b gene was confirmed in the proband, his twin brother and the mother, as shown in (Table 5). The SNV chr17:36104650 in the HNF1b gene was not found in the pool of 11 non-related individuals with congenital megaureter.
| Subjects | TBX3 S621R agC/agA |
GATA6 S156R agC/agA |
DHH A368V gCg/gTg |
LDB3 S437P Tcc/Ccc |
HNF1B G76C Ggc/Tgc |
|---|---|---|---|---|---|
| Proband | C/C | C/C | C/C | T/T | G/T |
| Proband Father | C/C | C/C | C/C | T/T | G/G |
| Proband Mother | C/C | C/C | C/C | T/T | G/T |
| Proband brother | C/C | C/C | C/C | T/T | G/T |
DISCUSSION
Studies with monozygotic twins can be decisive in the elucidation of the molecular mechanisms involved in the development of congenital diseases in humans, especially because, in contrast to other individuals, monozygotic twins share similar prenatal environments as well as a common genetic background [13]. In this study, we identified 40,123 SNVs in the proband. The overall read quality and read depth obtained with the WES was considered satisfactory. Comparison between the proband and his brother yield 99.2% of genotype similarity in common SNVs regions, confirming their condition of monozygotic twins.
In humans, heterozygous mutations of the HNF1b gene may result in several kidney abnormalities, once it is directly involved in nephrogenesis [37, 38]. Currently several phenotypes are associated with variations in the HNF1b gene including the formation of cysts, oligomeganephronia, renal agenesis, renal hypoplasia, and familial juvenile hyperuricemic nephropathy [39-44]. Our results indicated the presence of a heterozygote SNV in the HNF1b gene, position chr17:36104650 [Ggc/Tgc], in the proband, his twin brother and mother. This SNV resulted in a high PolyPhen-2 [36] score suggesting that this variant may lead to a damaging change in protein structure. Unfortunately, this result was not replicated in a pool of 11 non-related individuals with congenital megaureter. Therefore, our findings, despite suggestive, did not allow us to confirm a role of the gene HNF1b in the pathogenesis of the megaureter.
Other authors have suggested that the HNF1b gene cooperates with other genes as Pax8 and Lim1, during early organogenesis, to the construction of a healthy functional kidney [37, 38, 45, 46]. The hypothesis of a complex regulatory mechanism involving the participation of more than one gene is corroborated by the observation that the SNV at the HNF1b gene was detected in heterozygosis in two healthy individuals, the mother and the brother of the proband. Other studies involving patients with CAKUT have also failed in isolating genetic monogenetic determinants [7, 13, 47-49]. Recently, Jin et al. [47] published a study investigating monozygotic twins discordant for congenital renal agenesis from a set of blood samples using high-throughput exome-capture sequencing to detect single-nucleotide polymorphisms [SNPs], copy number variations [CNVs], insertions and deletions [indels] and differentially methylated regions [DMRs]. In this study, no discordant SNPs, CNVs, or indels were confirmed. To explain these results, some studies have suggested that, despite the apparent environmental similarities shared by these individuals, the intrauterine milieu is not necessarily equal for both twins [50]. Monozygotic twins could have differences due to the number of cells allocated to each twin, the timing of the twinning process, the vascular distribution from the placenta and epigenetic factors [12]. This can be especially true in monochorionic twins, the term used to refer to twins sharing the same placenta [51].
Our search for a genetic trigger for the development of megaureter was marked for several methodologic limitations. In this study, investigation of somatic mosaicism or RNAs expression in the affected tissue was impossible, since the samples obtained during the bilateral surgical re-implantation of the ureters in the proband were not available. In addition, despite being a promising tool, the WES method is still under development. For this reason, WES still poses challenges regarding the establishement of a standard method for data analysis and for the interpretation of the results [52]. Consequently, the combination of other bioinformatics techniques [53, 54], including data from prior GWAS studies, may still be needed for identifying causative mutations.
In conclusion, our study was not able to prove the association of HNF1b gene with the pathogenesis of the congenital megaureter by using WES technique and Sanger validation. In spite of that, we believe that the study of genetic differences within discordant monozygotic twins may help to decipher the complex network beyond genetic variations in the development of congenital megaureter.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.



