All published articles of this journal are available on ScienceDirect.

A Case of Anti-Glomerular Basement Membrane Crescentic Glomerulonephritis in a Female Renal Allograft Recipient with Unknown Native Disease: Retrospective Molecular Confirmation of Alport Disease
Authors Info & Affiliations
Abstract
Anti-Glomerular Basement Membrane (anti-GBM) crescentic glomerulonephritis developing in an allograft is a rare phenomenon. A patient with Alport syndrome receiving a renal transplant is at risk of developing anti-GBM glomerulonephritis, due to the absence of normal COL4α3, COL4α4 and COL4α5 trimer of the collagen network. Two unique challenges with planning kidney transplant in such a patient include- ideal donor selection; and risk of developing anti-GBM nephritis. We report a case of post-transplant anti-GBM crescentic glomerulonephritis in a female recipient with unknown native kidney disease who was diagnosed with Alport disease when she presented with hematuria and proteinuria nearly 2 years postrenal transplant. Allograft outcome in our case was unfavourable, patient reaching end-stage kidney disease within 6-month of diagnosis. The patient remains on continuous ambulatory peritoneal dialysis and currently active on the deceased donor transplant waiting list.
1. INTRODUCTION
Anti-GBM crescentic glomerulonephritis in an allograft is very rare. It may be either due to de-novo anti-GBM antibody formation or secondary to Alport syndrome(AS). Etiologies of de-novo disease could be similar to those occurring in native kidney, which as smoking, exposure to hydrocarbon & organic solvents, bacterial and viral infections [1]. Patients with AS have genetic mutations affecting type IV collagen in the glomerular basement membranes and the renal allograft recipient develops pathogenic anti-GBM antibodies which are deposited along the glomerular capillary walls. The incidence of anti-GBM crescentic glomerulonephritis is reported to be 1.9% in patients with AS [2]. It was first described in 1962 by McCoy et al., who put forth the idea of allogeneic response to the antigens or epitopes present in the donor kidney [3]. Recurrent Anti-GBM crescentic glomerulonephritis in the graft is very unusual [4]. Here, we report a case of postrenal transplant anti-GBM crescentic glomerulonephritis in a female allograft recipient with an unknown native disease, later resolved to be AS.
2. CASE REPORT
A 17-year old female received a renal allograft transplant from her haplo-matched father in May 2012 for unknown native kidney disease. She presented with edema, anemia, proteinuria (2+), bland urinary sediments, negative Anti-Nuclear Antibodies (ANA) and bilateral small kidneys on ultrasonography. She did not receive any induction therapy. Her maintenance immunosuppressive medication included tacrolimus, mycophenolate mofetil and steroids. The postoperative period was uneventful.
5-month post-transplant, the patient was treated with valganciclovir for fever, cytopenia, and positive Cytomegalovirus (CMV) DNA-PCR. Her allograft function remained stable with serum creatinine of 1.0 mg/dl for the subsequent 20 months. Periodic routine urine tests were negative for blood, protein and decoy cells.
In January 2014, she developed haematuria along with symptoms of upper respiratory tract infection that was treated with oral erythromycin. Two weeks later hematuria recurred with development of edema and worsening of serum creatinine from 1.1mg/dl to 1.8 mg/dl. Urine examination showed 3+ proteinuria, numerous red blood cells and epithelial cells. On examination, allograft was non-tender, with resistive index 0.6 on ultrasonography, normal serum complements (C3, C4) and Antistreptolysin O antibody titres. ANA was positive (1:40, speckled). Allograft biopsy was performed.
Allograft biopsy showed 7 viable glomeruli with active circumferential crescents in 5 (Fig. 1). There was a mild degree of tubulointerstitial chronicity. Immunofluorescence staining showed strong linear IgG along the glomerular capillary base ment membranes and occasionally along distal tubular basement membranes (Fig. 2). A diagnosis of Anti-glomerular basement membrane nephritis with crescentic transformation was made.
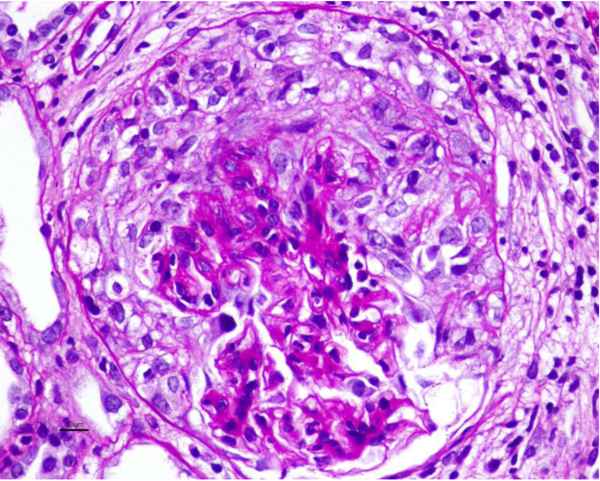
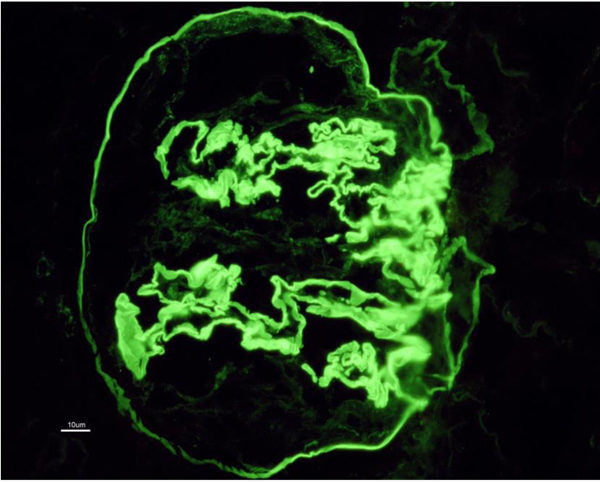
The patient was treated with pulse methylprednisolone (500 mg*3 doses). Plasmapheresis was initiated on day-2 whilst serum creatinine had increased to 2.2 mg/dl, and rituximab was added on day-3 of biopsy. 6 cycles of Plasmapheresis were given. The second dose of Rituximab 1 gm IV was given on day-11. Graft function marginally improved(serum creatinine 1.7 mg/dl) without any pulmonary symptoms.
Post-biopsy results (day-14), serum anti-GBM antibody was positive with titres of 91 (normal <12 IU/l); and negative anti neutrophil cytoplasmic antibody. Alport syndrome was suspected to be the native kidney disease in view of post-transplant anti-GBM crescentic glomerulonephritis. However, eye and ear examination were normal and the diagnosis of native disease was still unknown. Hence, the mutational study was ordered.
Clinical exome sequencing and variant analysis detected a high confidence homozygous A to G, C to T, and C to T substitution in the gene COL4 α3 on chromosome 2 at position 228113175, 2281135631 and 228173638, respectively; homozygous T to C and G to A substitution in the COL4 α4 on chromosome 2 at position 227872995 and 2279915832, respectively; heterozygous A to G substitution in the gene COL4 α5 on chromosome X at position 107865895.
In view of the positive anti-GBM antibody, plasmapheresis was continued. In all, 23 sessions were given. However, the graft function did not improve and deteriorated relentlessly over the next six months. A repeat graft biopsy was performed 6 months later to the first biopsy and revealed fibrous crescents in all 8 glomeruli (Fig. 3) with moderate tubule-interstitial chronicity. Immunofluorescence continued to reveal similar findings as first biopsy (Fig. (4). She is currently on continuous ambulatory peritoneal dialysis, waiting to receive a deceased donor kidney.
Parents of the index case were screened for routine urine analysis, eye and ear check-up. No abnormalities were found. Hence, further genetic studies were not carried.
3. DISCUSSION
Alport disease is a genetic condition affecting predominantly kidneys, ears and eyes. It is characterized by impaired production or assembly of type IV collagen network in the basement membrane due to mutations in COL4α3, COL4α4 or COL4α5 genes. Three genetic forms of AS have been identified: a] X-linked AS (85%) which is associated with mutations in COL4α5 gene, b] autosomal recessive AS (10%) where the patient is homozygous for mutations in COL4α3 and COL4α4 genes and c] autosomal dominant AS (5%) in which the patient has only one copy of mutations in either COL4α3 or COL4α4 genes (1). COL4α5 gene is located on the X chromosome and hence the full-blown disease is almost always seen in males. Autosomal mutations affect males and females equally.
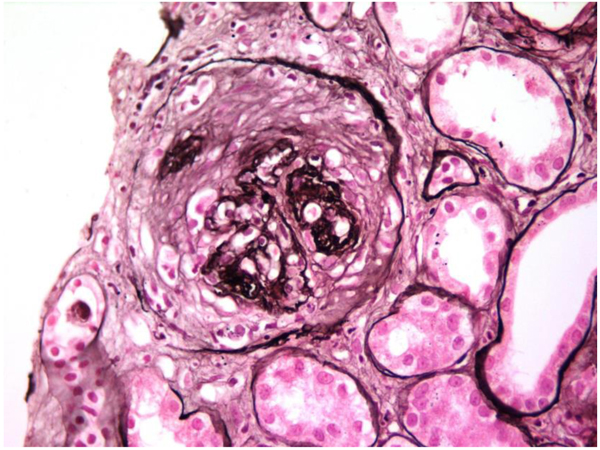
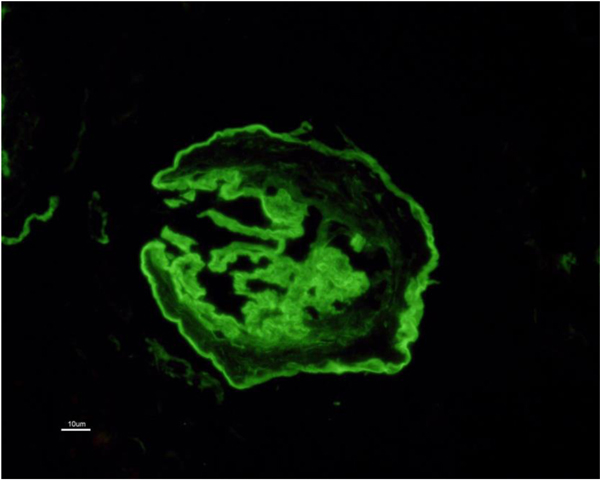
Affected patients manifest as hematuria and proteinuria in their childhood, sensorineural deafness and eye changes (lenticonus, keratoconus and cataract). Nephrotic syndrome is seen in 30-40% of cases. Renal histology reveals focal and segmental sclerosis in established cases, however, glomerular morphology could be very subtle in the early stages. Interstitial foamy histiocytes are frequently seen. Immunofluorescence results are negative with a panel of immunoglobulins (IgG, IgA, IgM), complements (C3, C1q) and light chains (kappa & lambda). Discontinuous staining with COL4α5 along glomerular basement membrane, Bowman’s capsule and distal tubular basement membranes are noted. On electron microscopy, thin glomerular basement membrane is seen in the early phase of the disease, which later on depicts a typical thick and thin membrane with lamellated and basket weave appearance [5]. The clinical course progresses with the slow deterioration of renal function and proteinuria if left untreated, and results in end-stage kidney disease (ESKD). Pathology at ESKD is non-diagnostic with chronic diffuse and global glomerulosclerosis with severe tubular atrophy accompanied by interstitial fibrosis. It is very difficult to distinguish from other etiologies, as multi-layering of basement membranes is commonly seen with ESKD from any cause. In such cases, genetic testing can help to establish the diagnosis [6]. In our patient, the kidneys were shrunken on initial presentation and hence pre-transplant biopsy was not done. A decision to proceed with kidney transplantation as definitive therapy in a young patient was taken as there was no other way to decipher the original reason for ESKD.
As patients with AS are deficient in collagen IV chains, development of pathogenic autoantibodies to the exposed epitopes in donor kidney during the post-transplant period is a recognized phenomenon [1]. Approximately 10% of the patients with Alport disease, who undergo a renal transplant, develop Anti-GBM antibodies without affecting graft function. Amongst them, only 1-2% of cases proceed to develop crescentic glomerulonephritis [3, 7]. Majority of such patients are males with X-linked Alport syndrome. On the contrary, anti-GBM disease in grafts of female recipients with Alport disease ise rarely described in the literature [8]. Development of graft dysfunction due to Anti-GBM nephritis ranges usually occurs within one year of transplantation. However, it may occur several years later too [9]. Rarely, immediate graft dysfunction is observed. Our patient manifested with hematuria at 21-month post-transplant, which is similar to reported cases in the literature.
Removal of pathogenic autoantibodies by plasma exchange and suppression of on-going production of autoantibodies with steroids and cytotoxic therapy remains the mainstay of management [10]. Initial improvement was noticed in our patient; however, she progressed to ESKD requiring dialysis support. Similarly, long term plasmapheresis and mycophenolate mofetil may attenuate illness, however, do not circumvent graft loss. Rituximab therapy has shown improvement in haematological parameters than renal function [11, 12]. Modification of histologic findings with Anti-T cell therapy was observed by Browne et al.; however, it failed to prevent graft loss [13].
Graft dysfunction due to Anti-GBM crescentic GN poses a high risk for recurrence in the subsequent transplants. Re-transplantation in Alport post-transplant Anti-GBM nephritis has been attempted in 3 cases by Browne et al. [13]. Authors have noticed a de-crescendo pattern in the form of shortened post-transplant duration for graft dysfunction /biopsy diagnosis, with successive transplantation. Alloantibodies to α5(IV)NC1 domain is the predominant antigen in this series undergoing re-transplantation. Among the various methodologies to detect serum autoantibodies to anti-GBM (western blot, ELISA and indirect immunofluorescence) – two out of three cases failed to detect using ELISA technique. The authors hypothesized Western blot as a more sensitive and specific method than standard ELISA [13].
Absence of eyes and ear abnormalities in our patient is intriguing and we do not have any explanation. However, we conclude the patient is more likely to have had autosomal recessive Alport syndrome, for the following reasons: a) homozygous mutation in COL4α3 and COL4α4 on chromosome 2; b) lack of hematuria in father which rules out X-linked AS and c) the early age of onset.
CONCLUSION
Alport disease is a genetic disease involving mutations in type IV collagen strands in the glomerular basement membrane. In our patient, the diagnosis of Alport disease was made retrospectively after the development of anti-GBM antibody crescentic glomerulonephritis in the renal allograft nearly 2 years post-transplant. The diagnosis was established after mutation studies in the recipient revealed the relevant mutations.
A high index of suspicion is the key to diagnosis, and anti-GBM crescentic glomerulonephritis should be considered as a cause of unexplained allograft dysfunction with hematuria in patients with ESKD of unknown etiology. Mutation study of the recipient is worth performing in such instances to confirm diagnosis as it would help to establish the cause for the allograft failure as well as help in choosing donors for a possible second transplant.
LIST OF ABBREVIATIONS
| ANA | = Antinuclear Antibody |
| ANCA | = Anti-neutrophil Cytoplasmic Antibody |
| Anti-GBM | = Anti-Glomerular Basement Membrane |
| AS | = Alport Syndrome |
| ASO | = Anti-Streptolysin O Titer |
| C3 | = Complement C3c |
| C4 | = Complement C4 |
| CMV | = Cytomegalovirus |
| DNA | = Deoxyribonucleic Acid |
| ELISA | = Enzyme-linked Immunosorbent Assay |
| GN | = Glomerulonephritis |
| PCR | = Polymerase Chain Reaction |
| AS | = Alport Syndrome |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Informed consent was obtained from the patient.
STANDARD FOR REPORTING
CARE guidelines and methodology were followed.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We sincerely thank Mrs. Tulasi Kumari, Mrs. Hema Nagaraj and Mrs. Bindushree for their outstanding technical support in Histopathology section.

