All published articles of this journal are available on ScienceDirect.

Aspirin Impairs Transport of Protective Osmolytes in Renal Inner Medullary Collecting Duct Cells
Authors Info & Affiliations
Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) represent one of the most common classes of medications used worldwide for treatment of pain and inflammation, and their nephrotoxic effects have been well documented. This study is focused on aspirin action on the normal adaptive responses of cells in the hypertonic renal inner medulla, using a cultured cell model. Following adaptation to hypertonic conditions, further treatment of both MDCK and IMCD3 cells for 24h with 1.0 mM aspirin produced significant inhibition of the transport systems for the osmolytes betaine and taurine. Caspase 3 was not activated by this treatment, indicating that transport inhibition was not due to apoptotic events. Acetaminophen and caffeine, common in analgesic mixtures, had a similar inhibitory effect. We conclude that interference with osmolyte accumulation by medullary cells may help to explain in part the nephrotoxicity due to long-term use of NSAIDs and analgesic formulations.
INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin and ibuprofen, are used worldwide as analgesics and antipyretics, and an estimated 6-7 billion $US is spent on them each year [1]. They are especially effective for treating inflammatory diseases (e.g. arthritis) through non-specific inhibition of cyclooxygenase (COX) enzymes which limits production of prostaglandins. The COX1 isoform appears to serve a constitutive housekeeping role, responsible for maintaining basic physiological functions, including cytoprotection of the gastric mucosa and control of platelet aggregation. In contrast, COX2 is induced by inflammatory mediators and mitogens and appears to play a key role in pathophysiological processes such as inflammation [2,3]. In the kidney, COX1 is highly expressed in the collecting duct, and low levels of COX1 are detected in interstitial cells, glomerular mesangial cells, and endothelial cells of arterioles. In contrast, COX2 is expressed predominantly in the renal medullary interstitial cells and in cortical thick ascending limb cells [4, 5]. The expression of COX2 in these cells also can be induced after a variety of physiological stresses, such as salt-loading and hypertonicity [2, 5]. One role may be to produce PGE2 during urinary concentration to regulate transport of water, salt and urea in collecting ducts. PGE2 also may improve survival of medullary cells during hypertonic stress [4]. Important side-effects of long-term NSAID therapy include injury to the gastrointestinal tract and papillary necrosis in the kidney. Selective COX2 inhibitors (e.g. vioxx, celecoxib) appear to cause similar problems [1]. NSAIDs may have direct effects on renal epithelial cells that express COX activity [6, 7], and also may drive proliferation and apoptosis through effects unrelated to COX inhibition [8, 9].
We are investigating if renal medullary injury by NSAIDs may be due in part to depletion of PGE2 and subsequent failure of the adaptive responses to the hypertonic inner medulla. Accumulation of osmolytes is important for cell survival in a hypertonic environment [10, 11], and atrophy of medullary tissue occurs when osmoprotective genes are not activated [12, 13]. The present study is focused on the action of aspirin on the transport of specific osmolytes, betaine and taurine, in renal medullary cells. Acetaminophen and caffeine, non-NSAIDS, were included in the study because of reports that heavy consumption of the former can contribute to renal failure. In addition, coformulations of aspirin and acetaminophen are popular in many cold and allergy preparations because the dose of each can be reduced. Caffeine is often added to these analgesic mixtures because it can increase efficacy by 50% [14, 15].
METHODS AND MATERIALS
Madin-Darby canine kidney (MDCK) and inner medullary collecting duct (IMCD3) cell lines, derived from the renal medulla of dog and mouse kidney respectively [16-18], were obtained from the American Type Culture Collection (Rockville, MD) and grown in monolayers in 24-well plates under standard culture conditions, as described in detail in our previous studies [19, 20]. The growth medium was DMEM/F12 (1:1) containing 10% bovine calf serum, 15 mM Hepes, 25 mM NaHCO3 (pH 7.4), 100 IU/ml penicillin and 100 μ g/ml streptomycin. Hypertonic growth medium was made by addition of NaCl crystals to normal isotonic growth medium to obtain a final osmolarity of 500 mOsm, as confirmed by direct measurement in an osmometer. When the cells reached confluent density in isotonic medium, some were switched to hypertonic 500 mOsm medium and allowed to equilibrate for 24 h prior to drug addition. Treatment with drugs was for 24 h in either isotonic or hypertonic growth medium. Osmolyte transport was assessed in the absence of drugs but immediately after the drug treatment period. Betaine/GABA transport (BGT1) activity was determined by measuring the initial rate (10 min uptakes) of Na+-dependent accumulation of [3H]GABA by cell monolayers, as in our previous studies [19, 20]. The osmolarity of all solutions used in the transport assays was matched to the previous growth medium by sucrose addition, as in earlier studies [19, 20]. Similarly, using 10 min uptakes, [3H]taurine was used as substrate for the Na+/taurine cotransporter [21], and [14C]methylaminoisobutyric acid (MeAIB) was used as substrate for the Na+ -dependent system A transporter [22]. Cells maintained in isotonic medium served as controls.
Preparation of whole cell lysates, gel electrophoresis and western blotting procedures were described in our previous studies [19, 20]. The blots were probed with a mixture of antibodies (1:1000 dilution) to both full-length and cleaved caspase 3 (Cell Signaling Technology, Beverly, MA), which were a kind gift from Dr Fred Pavalko in this department. Aspirin (salicylic acid), acetaminophen and caffeine were obtained from Sigma (St Louis, MO), and NS-398 was from Cayman Chemical (Ann Arbor, MI). Stock solutions were prepared in DMSO, ethanol or phosphate buffered saline, as appropriate, at 1000-fold working concentrations.
Data are presented as mean ± SD from 3-6 separate experiments. In each experiment the mean value was derived from triplicate determinations. Significant differences between groups were evaluated by ANOVA and Tukey’s test for multiple comparisons, using Instat v.3.06 software (GraphPad). A probability of p<0.05 was considered statistically significant.
RESULTS AND DISCUSSION
As expected, MDCK cells previously adapted to hypertonic (500 mOsm) growth medium for 24h showed a more than10-fold upregulation of BGT1 transport activity compared to isotonic controls (Fig. (1), aspirin absent). In this initial study the addition of aspirin (range 0.1 – 1.0 mM) to the hypertonic medium for a further 24h produced dose-dependent inhibition of BGT1 transport (Fig. 1). Inhibition was detected at 0.1 and 0.3 mM and appeared to be maximal at 1.0 mM because higher doses of 3.0 and 10.0 mM did not increase the degree of inhibition significantly (not shown). The inhibitory action at 1.0 mM compares well with the serum level of aspirin (1-2 mM) that is effective in treating rheumatoid arthritis [8]. Aspirin action on BGT1 transport in isotonic controls was not dose-dependent, although the percent inhibition at 1.0 mM was similar to that in hypertonic cells (Fig. 1). The therapeutic level of acetaminophen is about 0.1 mM and toxicity occurs when excessive consumption leads to serum levels that exceed 2.0 mM [8]. The plasma caffeine level after normal coffee consumption (1-2 cups) remains less than 0.1 mM [23] and may be fatal in the range 0.5 – 2.0 mM [8]. Higher levels of these drugs may occur locally in the renal medulla when water conservation is maximal. Based on these initial data and information, the standard protocol for comparing the different drugs was a 24 hr exposure to a concentration of 1.0 mM, using cells previously adapted to hypertonic stress (500 mOsm) for 24 hr.
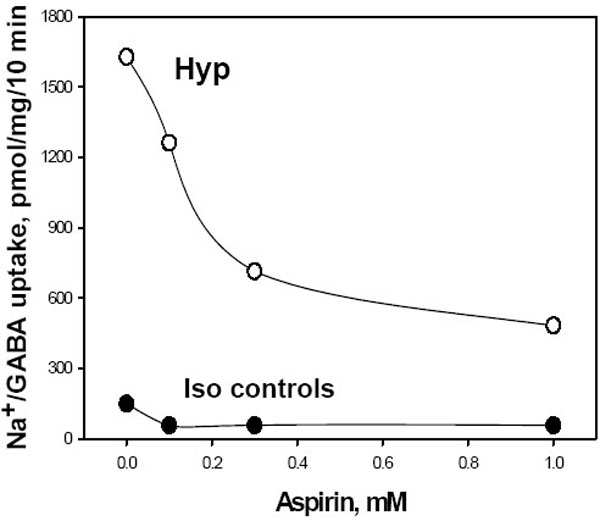
Dose dependent inhibition of BGT1 transport activity by aspirin after hypertonic upregulation. Cell monolayers were incubated for 24h in either hypertonic (Hyp, 500 mOsm) or normal isotonic (Iso) growth medium, followed by a further 24h at the same osmolarity and in the presence or absence of aspirin. BGT1 transport was then determined as Na+-dependent uptake of [3H]GABA, after a 10 min incubation, and corrected for cell protein. Data are mean of two experiments, each analyzed in triplicate.
Two different medullary cell lines were compared in parallel in order to focus on consistent effects of drug treatment and to avoid cell-specific responses. Control IMCD3 cells in normal isotonic growth medium showed no response to drugs. In contrast, IMCD3 cells adapted to hypertonic medium showed significant inhibition of BGT1 after 24hr exposure to aspirin, acetaminophen and caffeine at 1.0 mM (Fig. (2), upper panel). The response of MDCK cells was similar, namely all drugs inhibited BGT1 transport after hypertonic adaptation but there was no significant change in the isotonic controls (Fig. (2), lower panel).
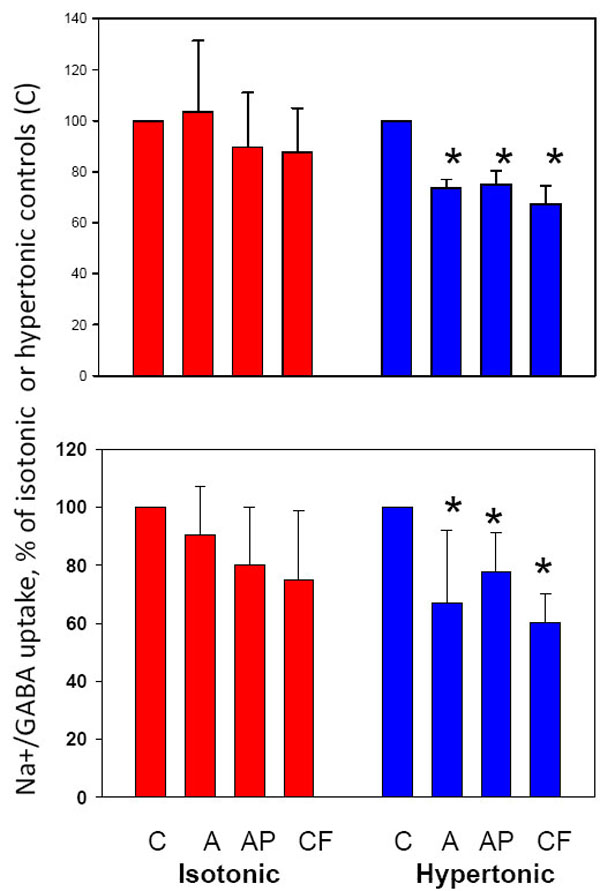
Inhibition of BGT1 transport in IMCD3 and MDCK cell monolayers by aspirin, acetaminophen and caffeine at 1.0 mM. All treatments produced significant inhibition of BGT1 transport in IMCD3 (upper panel) and MDCK (lower panel) cells adapted to hypertonic medium but isotonic controls were not altered significantly. *Significantly different (p<0.05) from appropriate untreated controls (C). Data are mean ± SD from 3 (IMCD3) or 5-6 (MDCK) experiments. Absolute values for BGT1 transport in C groups of IMCD3 were 59±13 (isotonic) and 161±30 (hypertonic) pmol/mg/10 min, and values for C groups of MDCK were 198±35 (isotonic) and 1356±336 (hypertonic) pmol/mg/10 min. A, aspirin; AP, acetaminophen; CF, caffeine.
Transport of taurine, another osmolyte, also was inhibited by aspirin and acetaminophen in IMCD3 cells following hypertonic stress (Fig. 3). NS-398, a specific inhibitor of COX2 [2, 5, 8] reproduced the inhibitory action of aspirin on taurine transport (Fig. 3) and BGT1 (not shown), suggesting that prostaglandins may be important for maintaining upregulation of osmolyte transport during hypertonic stress. NS-398 was used at 0.01 mM final concentration which does not affect COX1 and was previously shown to inhibit PGE2 production in IMCD3 cells [8]. As with BGT1 transport (Fig. 2) the drugs had no effect under isotonic conditions (Fig. 3), suggesting that hypertonicity may sensitize the cells to drug action on these transport systems.
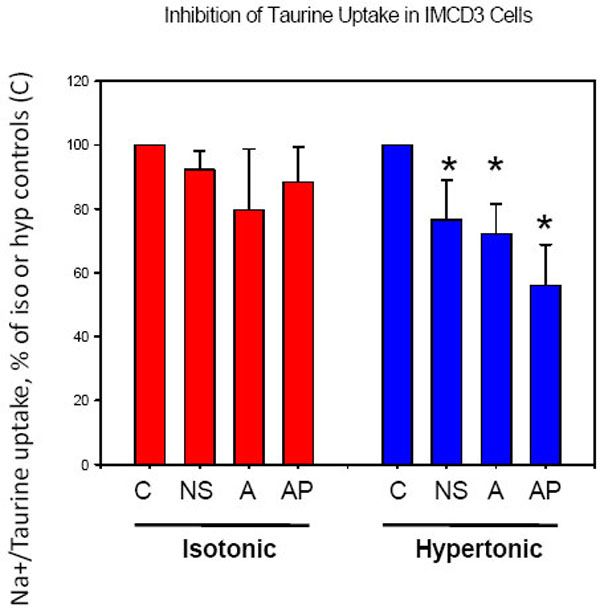
Inhibition of taurine transport in IMCD3 cells by a COX2 inhibitor, aspirin and acetaminophen. All treatments inhibited taurine uptake in cells adapted to hypertonic stress, but there was no effect on isotonic control cells. *Significantly different (p<0.05, n=4) compared to untreated group C. Absolute values for taurine transport in group C were 672±223 (isotonic) and 1340±240 (hypertonic) pmol/mg/10 min. NS (NS-398) was present at 0.01mM, and A and AP were 1.0 mM.
In contrast to osmolyte transporters, the system A transporter for amino acids showed no significant response to NS-398, aspirin and acetaminophen in both IMCD3 and MDCK cells (Fig. 4). This indicates that inhibition of BGT1 and taurine transport by these drugs was not simply a non-specific effect due to disruption of the transmembrane sodium gradient, which is the driving force for these transport systems.

The system A amino acid transporter was not inhibited by NS-398, aspirin or acetaminophen. System A transport in IMCD3 (upper) and MDCK (lower) cells was determined as Na+- dependent uptake of [14C]MeAIB, and was not altered by drug treatment. See Fig. (3) for other details.
Hypertonic stress alone or in combination with aspirin treatment did not activate caspase 3 in IMCD3 and MDCK cells. Western blotting of whole cell lysates detected only the
full-length inactive caspase 3 at 35 kDa (Fig. 5A), indicating that apoptosis was not induced. Lysate samples from osteoblastic cells were used as controls to validate the western technique. The activated 17kDa fragment of caspase 3 was detected in lysates from cells made apoptotic by addition of tumor necrosis factor-α and cycloheximide, as expected from previous reports [24, 25], but was absent from untreated controls (Fig. 5B). In addition, the drug-treated kidney cells showed no evidence of rounding or detachment. Taken together, these findings indicate that inhibition of BGT1 and taurine transporters by 1.0 mM aspirin treatment for 24h was not an indirect result of apoptotic events. Aspirin at higher concentrations and longer exposure was shown previously to induce DNA damage and apoptosis, especially in rapidly proliferating IMCD3 cells [8,9].
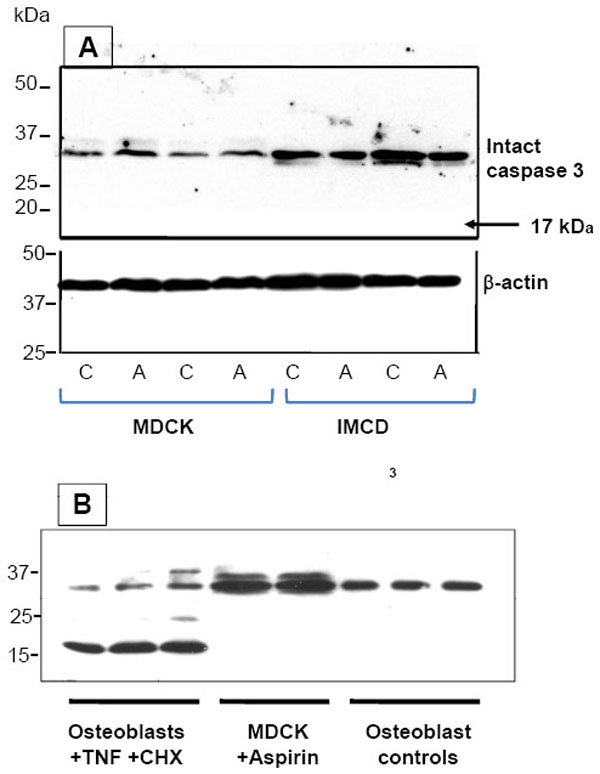
Aspirin did not activate caspase 3 in either MDCK or IMCD3 cells. (A) Western blotting of whole cell lysates detected only full-length inactive caspase 3 (35kDa). No activated cleaved fragments were present. Blots were probed with a mixture of antibodies to both intact and cleaved caspase 3. All cells were adapted to hypertonic growth medium for 24h prior to a further 24h incubation in the presence (A) or absence (C) of 1.0 mM aspirin. (B) Lysates from mouse osteoblastic (MC3T3-E1) cells were used as controls. When these cells were made apoptotic by 4h exposure to tumor necrosis factor-α (TNF, 5 ng/ml)) and cycloheximide (CHX, 10 ng/ml), a 17kDa fragment of activated caspase 3 was detected, as expected from a previous study [25]. No activated fragments were detected in untreated controls or in aspirin-treated MDCK cells.
This report of consistent findings in two different lines of renal medullary cells is the first description of impairment of renal osmolyte transport by NSAIDs and analgesics. The potential mechanisms have not been determined although non-specific toxicity and apoptosis can be ruled out. Over-the-counter aspirin is sold as acetylsalicylic acid which is rapidly converted to salicylic acid after ingestion [8], and is the form tested here in vitro. The action of aspirin was reproduced by a COX2 specific inhibitor, suggesting that aspirin may act via inhibition of COX2 and possible depletion of prostaglandins important for maintaining the adaptation to hypertonic stress. Further studies are needed to confirm this. Caffeine inhibition of osmolyte transport may be related to its action to mobilize intracellular stores of Ca2+ in many cells including MDCK [26, 27]. We have previously shown that an increase in intracellular Ca2+ is associated with prompt endocytic retrieval of BGT1 protein from the plasma membrane [20]. The mechanism of acetaminophen inhibition remains to be determined but it is possible to speculate based on studies of acetaminophen action in the liver. These have led to the proposal that the metabolites of acetaminophen may eventually deplete intracellular glutathione which will be followed by onset of oxidative stress [9, 28]. A similar response in cells of the renal medulla would be very significant because we have previously observed a direct inhibitory action of oxidative stress on specific plasma membrane transport systems in cultured kidney cells [29].
In summary, interference with the ability of medullary cells to accumulate osmolytes may contribute to certain nephrotoxic effects in vivo, such as papillary necrosis [30], that result from long-term use of NSAIDs and analgesic mixtures containing acetaminophen and caffeine. The hypertonic environment in the inner medulla in vivo also may sensitize the cells to the action of these drugs. Additional toxic effects, as reported elsewhere, may occur in vivo due to concentration of these drugs in the renal inner medulla during antidiuresis.

