All published articles of this journal are available on ScienceDirect.

Mediastinal-Renal Syndrome: A Rare Manifestation of Granulomatosis with Polyangiitis
Authors Info & Affiliations
Abstract
Granulomatosis with polyangiitis (GPA) is a distinct systemic disorder of obscure etiology characterized by necrotizing granulomatous inflammation and pauci-immune small-vessel vasculitis. Anti-neutrophil cytoplasmic antibodies (ANCA) appear to play an important role in the pathogenesis of GPA. Herein, we report a rare case of GPA presenting with ANCA-associated pauci-immune necrotizing and crescentic glomerulonephritis accompanied by a mediastinal mass and mediastinal lymphadenopathy in the absence of pulmonary parenchymal involvement. Furthermore, we describe the effects of plasmapheresis and concomitant immunosuppression on renal and mediastinal disease.
INTRODUCTION
Formerly known as Wegener’s granulomatosis, granulomatosis with polyangiitis (GPA) is a distinct systemic disorder of obscure etiology characterized by necrotizing granulomatous inflammation and pauci-immune small-vessel vasculitis [1-4]. It frequently affects the upper and lower respiratory tract, sinuses, ears, eyes, kidneys, joints, skin, nervous system, and gastrointestinal tract. It also causes constitutional symptoms such as fatigue, unexplained weight loss, and fever. Anti-neutrophil cytoplasmic antibodies (ANCA) are present in 80%-90% of patients with GPA and appear to play an important role in its pathogenesis [2]. We describe a patient with GPA that was first evident in the mediastinum. Evolution of rapidly progressive glomerulonephritis led to therapy that reversed both mediastinal and renal manifestations.
CASE RESPORT
A 48-year-old man presented to this hospital with severe epigastric and substernal chest pain as well as shortness of breath for one day. The past medical history was notable for chronic lower back pain, pneumonia, and a recent “ear infection”. His medications included ciprofloxacin, acetaminophen, hydrocodone, and cyclobenzaprine (a muscle relaxant). The patient was a former carpenter and had a 35-pack-year history of smoking cigarettes. The review of systems was remarkable for unintentional weight loss (7 kg), night sweats, and subjective fevers within one month prior to presentation. On examination, the patient was an alert Caucasian man in no acute distress. The temperature was 37.2°C, the blood pressure 110/78 mm Hg, the pulse 86 beats per minute, the respiratory rate 12 breaths per minute, and the oxygen saturation 100% while the patient was breathing ambient air. On examination, the lungs, heart, abdomen, extremities, and skin were normal. The results of laboratory studies are shown in Table 1. A complete blood count showed mild leukocytosis, neutrophilia, eosinophilia, and iron deficiency anemia (transferrin saturation 6%). A urinalysis revealed hematuria and low-grade proteinuria. The level of serum albumin was low at 2.0 mg/dl.
Laboratory Data
| Variable | Reference Range, Adults | On 1stAdmission | On Discharge | On 2nd Admission |
|---|---|---|---|---|
| Sodium (mmol/L) | 135–145 | 140 | 140 | 134 |
| Potassium (mmol/L) | 3.4–4.8 | 4.3 | 4.0 | 4.6 |
| Chloride (mmol/L) | 99–109 | 104 | 107 | 102 |
| Carbon dioxide (mmol/L) | 21–30 | 28 | 26 | 26 |
| Urea nitrogen (mg/dl) | 7–22 | 7 | 14 | 34 |
| Creatinine (mg/dl) | 0.8–1.4 | 0.9 | 1.8 | 4.4 |
| Glucose (mg/dl) | 65–99 | 118 | 93 | 110 |
| Calcium (mg/dl) | 8.6–10.3 | 8.9 | 7.9 | |
| Albumin (g/dl) | 3.5–5.2 | 2.0 | 2.0 | |
| Globulin (g/dl) | 2.6-4.6 | 2.6 | 2.8 | |
| Aspartate transaminase (IU/L) | 5-45 | 24 | 57 | |
| Alanine transaminase (IU/L) | 5-60 | 15 | 43 | |
| Alkaline phosphatase (IU/L) | 30-115 | 99 | 101 | |
| Bilirubin, total (mg/dl) | 0.1-1.2 | 0.5 | 0.6 | |
| Hemoglobin (g/dl) | 13.6–16.7 | 13.0 | 9.8 | 9.1 |
| Hematocrit (%) | 40.0–49.0 | 40.4 | 30.3 | 28.0 |
| Mean corpuscular volume (µm3) | 82-93 | 89 | 88 | 85 |
| White-cell count (x103 per mm3) | 4.0–9.0 | 10.6 | 8.4 | 10.4 |
| Differential count (%)) | ||||
| Neutrophils | 41–67 | 71 | 62 | 83 |
| Lymphocytes | 28–42 | 14 | 13 | 13 |
| Monocytes | 4–9 | 8 | 10 | 10 |
| Eosinophils | 0–5 | 7 | 14 | 12 |
| Basophils | 0–1 | 0 | 0 | 1 |
| Platelet count (x103 per mm3) | 130–350 | 339 | 316 | 432 |
| Urinalysis | ||||
| Color | Yellow | Amber | Yellow | Yellow |
| Turbidity | Clear | Clear | Hazy | Hazy |
| pH | 4.6–7.8 | 5.5 | 5.5 | 6.5 |
| Specific gravity | 1.001–1.030 | 1.020 | 1.015 | 1.010 |
| Glucose | Negative | Negative | Negative | Negative |
| Ketones | Negative | Negative | Negative | Negative |
| Bilirubin | Negative | Negative | Negative | Negative |
| Blood | Negative | 3+ | 3+ | 3+ |
| Albumin | Negative | 1+ | 2+ | 2+ |
| Nitrites | Negative | Negative | Negative | Negative |
| Leukocyte esterase | Negative | Negative | Trace | Negative |
| White blood cells (per hpf) | 0-2 | 0-5 | 0-5 | 5-10 |
| Red blood cells (per hpf) | 0-2 | 20-50 | 10-20 | Numerous |
Values out of reference range are bold.
The patient was admitted to this hospital for the evaluation and treatment of epigastric and substernal chest pain. The patient ruled out for myocardial infarction. An upper gastrointestinal endoscopy revealed several localized erosions in the stomach and duodenum while colonoscopy was normal. The patient was diagnosed with erosive gastritis and placed on esomeprazole. A computed tomographic angiography revealed a non-enhancing soft-tissue mass in the aortopulmonary window and enlarged mediastinal lymph nodes, including a 2.3 cm left suprahilar lymph node and a 1.7 cm right hilar lymph node (Fig. 1A). In addition, there was poor opacification of the right subclavian vein along with extensive collateral circulation in the right chest wall consistent with central venous stenosis or occlusion (Fig. 1). The patient underwent video-assisted thoracoscopic surgery and sampling of the mediastinal lesions. The histological examination demonstrated fibrous connective tissue containing an inflammatory cell infiltrate comprised of mononuclear inflammatory cells, plasma cells, eosinophils, and occasional mast cells suggestive of fibrosing mediastinitis (Fig. 2A, B). Mononuclear and polymorpho-nuclear inflammatory cells, as well as nuclear debris, were found in the wall of several small blood vessels indicative of small-vessel vasculitis (Fig. 2C, D). Immunostaining revealed numerous T lymphocytes (CD3+) scattered throughout the lesion (Fig. 2E). In addition, B lymphocytes (CD20+) were present in small irregular aggregates and individually dispersed throughout the lesion (Fig. 2F). Immunostaining for CD3, CD15, CD20, CD30, cytokeratin 5/6, and calretinine did not suggest aneoplastic process. A Fite’s stain for acid-fast bacili and a Grocott's methenamine silver stain for fungal organisms were negative. Laboratory tests for a possible Histoplasma capsulatum infection (serology and urinary antigen testing) were negative.
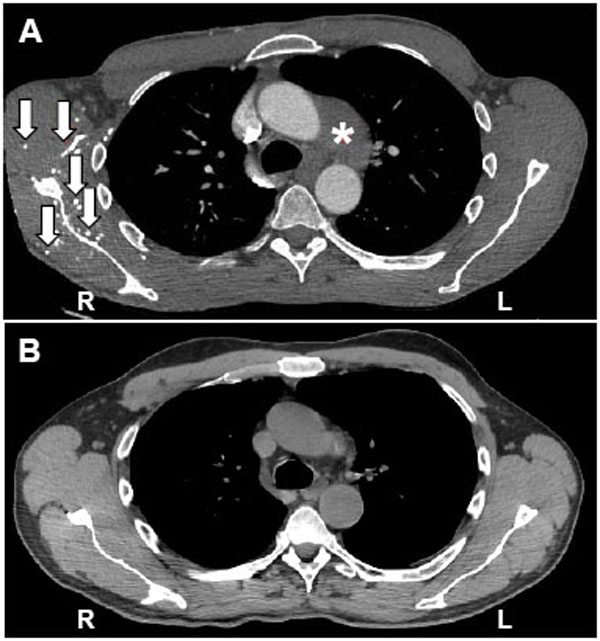
Computed tomography scans of the chest. A nonenhancing soft-tissue mass in the aortopulmonary window (*) is seen on a CT angiogram of the chest (A). Extensive venous collateral circulation in the chest wall (arrows) indicates central venous stenosis or occlusion (A). A decrease in the size of softtissue mass in the aortopulmonary window four months after the initiation of immunosuppressive therapy on a non-enhancing highresolution CT scan of the chest (B). There is no evidence of fibrosis in the mediastinal fat (B).
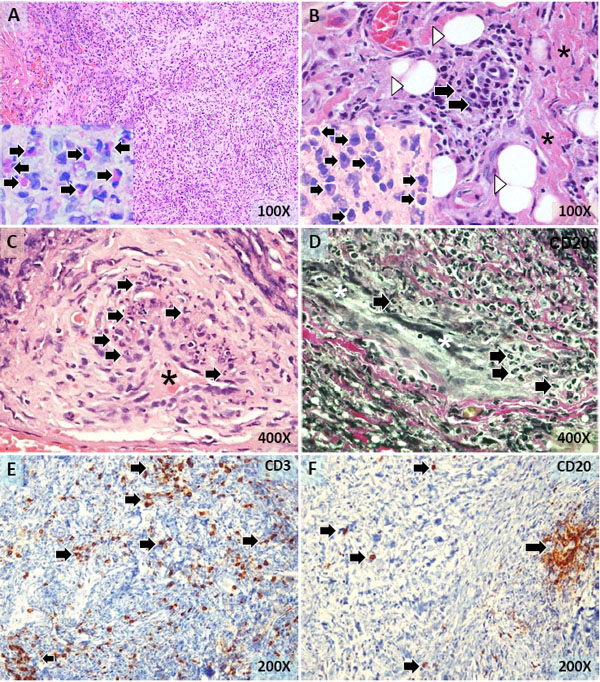
The histology of the mediastinal mass. Fibrous connective tissue containing numerous mononuclear and polymorphonuclear inflammatory cells (A). Numerous eosinophils (arrows in the insert) were present (A). A focus of inflammatory cell infiltration (arrows),fibrous connective tissue (*), and scattered residual fat cells (arrowheads) (B). Numerous plasma cells (arrows in the insert) were observed (B). Small blood vessels (*) with mononuclear and polymorphonuclear inflammatory cells (arrows) in the wall indicative of small-vessel vasculitis (C,D). Numerous T lymphocytes (arrows) scattered throughout the lesion (E). B lymphocytes forming a small irregular aggregate (arrowhead) and individually dispersed (F). Tissue sections were stained with hematoxylin-eosin (A-C) and elastic Van Gieson (D). Tissue sections were also immunostained for CD3 (E) and CD20 (F). Magnifications are as indicated.
While the work-up of mediastinal lesion was in progress, the level of serum creatinine rose to 1.8 mg/dl. The fractional excretion of sodium was found to be less than 1%. Renal insufficiency was attributed to poor oral intake, and nephrology consultation was not requested. On the 12th hospital day, the patient requested that the evaluation and treatment of his illness to be continued on an outpatient basis. He was discharged from the hospital against medical advice.
One week after discharge from the hospital, the patient returned to the emergency room complaining of fatigue, malaise, loss of appetite, hypersomnia, and weight loss. On examination, the patient was alert and in no acute distress. The temperature was 36.8°C, the blood pressure 119/69 mm Hg, the pulse 80 beats per minute, the respiratory rate 10 breaths per minute, and the oxygen saturation 96% while the patient was breathing ambient air. On examination, the conjunctiva of the right eye was red and swollen. An erythematous papular rash and multiple excoriations were noted on the trunk. The rest of the physical examination was unremarkable. The results of laboratory data revealed mild leukocytosis, neutrophilia, eosinophilia, and anemia (Table 1). Serum levels of creatinine and urea nitrogen were 4.4 mg/dl and 34 mg/dl, respectively (Table 1). A urinalysis demonstrated hematuria and proteinuria. Microscopic examination of the urine sediment revealed numerous dysmorphic red blood cells, several red blood cell casts, and occasional white blood cells (Table 1). The concentrations of protein and creatinine in a random sample of urine were 207 mg/dl and 92.5 mg/dl, respectively. The urine protein-to-creatinine ratio indicated 2.2 grams protein excretion per day. The renal ultrasonography demostrated mildly enlarged kidneys measuring 13.6 cm in length with no hydronephrosis, stones, or masses. Tests for anti-nuclear antibodies, anti-glomerular basement membrane antibodies, viral hepatitis (hepatitis B surface antigen, hepatitis B surface antibody, hepatitis B core antibody, hepatitis C antibody), and HIV were all negative. Rheumatoid factor was elevated at 157 IU/ml (normal: < 20 IU/ml). Serum levels of complements C3 and C4 were normal. Indirect immunofluorescence microscopy demon-strated a strong reaction for anti-neutorphil cytoplasmic anti-bodies (ANCA) with a cytoplasmic distribution (cANCA). Neither perinuclear (pANCA) nor atypical ANCA was observed. The enzyme-linked immunosorbent assay (ELISA) revealed antibodies against proteinase 3 (anti-PR3) at 42.1 U/ml (normal ≤ 3.5 U/ml). Anti-myeloperoxidase (anti-MPO) antibodies were not detected. The patient was diagnosed with episcleritis by the ophthalmology service and treated with prednisolone 1% eye drops. In addition, a rapidly progressive glomerulonephritis was diagnosed on clinical grounds leading to a renal biopsy. The tissue sample contained 36 glomeruli, 60% of which showed segmental fibrinoid necrosis and cellular crescents (Fig. 3A-C). There was no endocapillary or mesangial glomerular proliferation. The interstitium demonstrated a mononuclear inflammatory cell infiltrate, rare eosinophils, and edema (Fig. 3). The tubular epithelial cells demonstrated focally degenerative/regenerative changes (Fig. 3). There was no evidence of arteritis. Immunofluorescence microscopy did not reveal IgG, IgA, IgM, C3, C4, C1q, kappa or lambda light chain deposition in glomeruli. A histological diagnosis of pauci-immune necrotizing and crecentric glomerulonephritis was rendered.
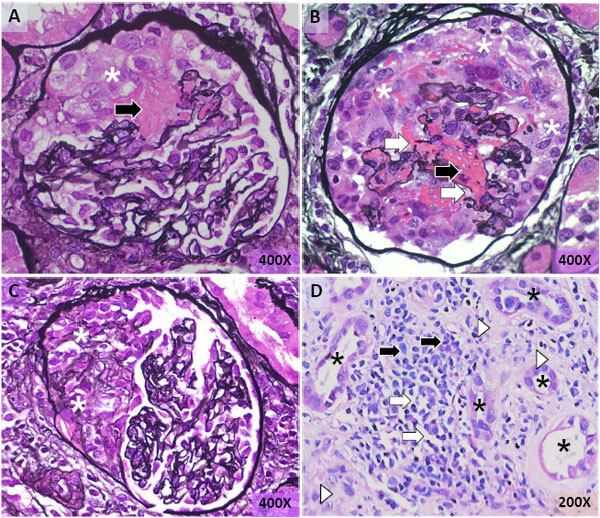
The histology of the kidney. A glomerulus showing accumulation of cells (*) and fibrinoid material (arrow) in the Bowman’s space forming a cellular crescent (A). A glomerulus showing a circumferential cellular crescent (*), segmental fibrinoid necrosis of the glomerular tuft (black arrow), and focal disruption of glomerular capillary walls (white arrows) (B). A glomerulus demonstrating a fibro-cellular crescent (*) (C). The interstitium showing mononuclear inflammatory cell infiltration (black arrows), edema (white arrows), and fibrosis (arrowheads). Separated by the expanded interstitium, the tubules (*) showing regenerative and degenerative changes in the epithelium (D). Tissue sections were stained with methenamine silver-periodic acid-Schiff (A-C) and hematoxylin-eosin (D). Magnifications are as indicated.
The patient was diagnosed with ANCA-associated small-vessel vasculitis involving the kidneys (necrotizing and crescentic glomerulonephritis), mediastinum (soft-tissue mass and lymphadenopathy), pleura (pleuritis and pleural effusion), right eye (episcleritis), and possibly right ear (“ear infection”), consistent with GPA. He was started on plasmaphoresis (a total of seven treatments), cyclophospha-mide 1250 mg intravenously, and methylprednisolone 1.0 g intravenously daily for 3 days followed by prednisone 60 mg orally daily. Soon after the initiation of the treatment, the patient’s symptoms improved and his renal function stabilized. He was discharged home on the 10th hospital day, at which time serum level of creatinine had declined to 1.6 mg/dl and anti-PR3 antibodies were undetectable. Following discharge from the hospital, the patient was treated with intravenous cyclophosphamide 1250 mg every months for a total of six months, prednisone 60 mg daily tapered to 5 mg daily over 3 months, and sulfamethoxazole-trimethoprim 400/80 mg once a day. After completion of cyclophos-phamide therapy, the patient was started on azathioprine 50 mg orally twice a day. Four months after the initiation of immunosuppressive therapy, a high-resolution CT scan of the chest demonstrated resolution of the mediastinal lymphadenopathy. In addition, there was no evidence of pulmonary or mediastinal fibrosis and the mass had resolved (Fig. 1B).
DISCUSSION
Herein, we report a rare case of GPA presenting with ANCA-associated pauci-immune necrotizing and crescentic glomerulonephritis accompanied bya mediastinal mass and mediastinal lymphadenopathy in the absence of pulmonary parenchymal involvement. Furthermore, we report on the efficacy of plasmapheresis and concomitant immunosuppres-sion in the treatment of the renal disease as well as mediastinal pathology in this patient.
GPA frequently involves the lungs (85%-90%) and the kidneys (80%-85%) [1, 2]. However, hilar lymphadenopathy (2%) and mediastinal mass (1%) are distinctly rare in this entity [5]. It has been reported that the histopathological examination of the mediastinal lesions in GPA demonstrates various degrees of granulomatous inflammation, necrosis, and/or vasculitis [5]. It is also important to mention that the mediastinal involvement in GPA is usually accompanied by pulmonary parenchymal disease [5]. As an example, an anterior mediastinal mass and subcarinal lymphadenopathy was reported in a 34-year-old man with cANCA-associated GPA who developed pulmonary hemorrhage and glomerulonephritis [6]. In that patient the histological examination of the mediastinal mass revealed granulomatous inflammation and necrotizing vasculitis [6]. However, it is quite rare to find absence of pulmonary parenchymal disease in patients having GPA with mediastinal involvement. In this context, a mediastinal mass due to necrotizing granulo-matous inflammation and vasculitis has also been reported in a 42-year-old man with GPA who later developed necrotizing glomerulonephritis [7]. Similar to our case, this 42 year-old-man did not demonstrate any evidence of pulmonary parenchymal disease [7]. In the current case, the histopathological features of the mediastinal lesion are distinct. While an acute vasculitis affecting small blood vessels was present, necrotizing granulomatous inflam-mation was not a dominant histopathological feature. Instead, a mixed inflammatory cell infiltrate comprised of T and B lymphocytes, eosinophils, plasma cells, and occas-ional mast cells in a fibrotic background was suggestive of fibrosing mediastinitis.
Fibrosing mediastinitis (FM) is a rare condition of obscure etiology characterized by a locally invasive fibro-inflammatory process within the mediastinum [8, 9]. FM is associated with substantial morbidity and mortality as a result of the compression and/or invasion of mediastinal blood vessels, heart, trachea, bronchial tree, and esophagus [8, 9]. The extensive venous collateral circulation observed on the CT scan of our case provides evidence for central venous stenosis or occlusion (Fig. 1A). A number of precipitating factors have been associated with FM, including fungal infections (especially histoplasma capsulatum), tuberculosis, and sarcoidosis [8]. Although the pathogenesis of FM in the majority of patients remains obscure, immune-mediated mechanisms appear to play an important role [8, 9]. Histologically, the accumulation of collagen resulting in dense fibrosis (sclerosis) is considered the hallmark of FM. However, mediastinal fibrosis appears to represent the advanced stage of a dynamic inflammatory process with evolving histologic appearance over time [10]. In fact, at an early stage during the development of FM, the accumulation of spindle-shaped cells, lymphocytes, plasma cells, eosinophils, mast cells, and thin-walled blood vessels occurs in a fibromyxoid connective tissue. This resembles the histological pattern observed in the case reported here. However, vasculitis is not a feature of FM. “Sclerosing mediastinitis” characterized by necrotizing granulomatous inflammation and vasculitis, which was diagnosed at necropsy of a 9-year-old girl with granulomatous and necrotizing glomerulonephritis and diffuse pulmonary hemorrhage due to acute capillaritis, appears to have been a rare manifestation of GPA, as described above [11].
In conclusion, the patient reported here had a rare presentation of GPA with ANCA-associated pauci-immune necrotizing and crescentic glomerulonephritis and mediastinal involvement. The patient responded very well to plasmapheresis and immunosuppression with glucocorticoids and cyclophosphamide resulting in substantial improvement in the renal function and mediastinal lesions. In addition, anti-PR3 antibodies became undectable at the time of discharge from the hospital. The use of plasma exchange in the treatment of GPA involving the kidneys is traditionally reserved for patients with a serum creatinine greater than 5.7 mg/dl. However, a recent study demonstrated that patients with a serum creatinine >2.85 mg/dl also benefited from addition of plasmapheresis to an immunosuppressive regimen consisting of prednisolone and cyclophosphamide [12]. A combination therapy consisting of glucocorticoids and cytotoxic agents were also reported effective in the treatment of mediastinal involvement in GPA [5]. In summary, the mediastinal-renal syndrome may be a rare manifestation of GPA. In our case, it responded well to immunosuppression with glucocorticoids and a cytotoxic agent.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

