All published articles of this journal are available on ScienceDirect.

Antiphospholipid Antibodies and APS Nephropathy
Abstract
The presence of pathogenic antiphospholipid antibodies (aPL) is the characterizing feature of the antiphospholipid syndrome (APS), mediating the recurrent pregnancy loss and thrombosis typical of the disease through its action on various antigenic targets. APS nephropathy is the characteristic clinico-athological manifestation of renal involvement in APS and occurs as a result of vaso-occlusive disease in the intrarenal vasculature. The typical clinical features and morphological lesions of APS nephropathy have been well characterized and several studies have established a link between these features and the presence of various aPL. In this review, we outline the proposed pathophysiological mechanisms of aPL-mediated thrombosis, the characteristic clinical and morphological features of APS nephropathy and the evidence linking aPL action to the occurrence of APS nephropathy.
INTRODUCTION
Antiphospholipid syndrome (APS) is an autoimmune disorder characterized clinically by arterial and venous thrombosis and recurrent miscarriage and late pregnancy loss in the presence of antiphospholipid antibodies (aPL). These aPL include a heterogenous group of antibodies, anticardiolipin (aCL) and anti-β2glycoprotein I (anti-β2GPI) antibodies and lupus anticoagulant (LA), and are responsible for the clinical manifestations noted in the disease through their action on a myriad of phospholipid (PL), PL-protein complex and PL-binding protein antigenic targets with which they interact [1]. APS can be classified as primary APS (PAPS), occurring in the absence of a related connective tissue disease (CTD) such as systemic lupus erythematosus (SLE) or as secondary APS (SAPS), occurring in their presence. The syndrome was first described in a subset of patients with SLE and related CTDs who had abnormal LA tests [2]. The pathophysiological mechanisms that contribute to this procoagulant phenotype include aPL-mediated activation of platelets, monocytes and endothelial cells (ECs) and aPL-induced perturbation of the natural anticoagulant and fibrinolytic systems [3].
The increased propensity for thrombosis may involve the vasculature of any organ in the body. Renal involvement represents one of the most important features of this syndrome, occurring in 2.7% of patients with APS [4]. However, the true incidence of renal involvement in APS is likely higher due to the fact that renal biopsies have often been discouraged because of the frequent occurrence of anticoagulant therapy, thrombocytopenia, systemic hypertension and a concern that biopsy-related complications were more common in APS patients [5]. Indeed, renal involvement has been reported to occur in up to 78% cases in patients with catastrophic APS, a more severe form of APS characterized by multi-organ involvement and widespread acute micro-vascular occlusions [6].
The manifestations of renal disease in APS reflect the progression of thrombosis along the kidney vascular bed: the main renal arteries and veins, intra-renal arteries, veins, arterioles, venules and glomerular capillaries. Glomerulonephritis has also been described [6]. The characteristic presentation of renal involvement in APS has been termed APS nephropathy, which denotes the parenchymal and vascular damage to kidneys caused by aPL-mediated acute or chronic intraluminal and intramural occlusive lesions in arterioles, interlobular arteries and glomeruli [5, 6]. Other causes of renal microangiopathy should be ruled out, including diabetes mellitus (DM), thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), cyclosporine use and systemic sclerosis. In recent years, the typical morphological lesions of APS nephropathy have been well characterized and several studies have established a link between these features and the presence of various aPL [7]. In this review, we outline the proposed pathophysiological mechanisms of aPL-mediated thrombosis, the characteristic clinical and morphological features of APS nephropathy and the evidence linking aPL action to the occurrence of APS nephropathy.
PATHOPHYSIOLOGICAL MECHANISMS OF THROMBOSIS IN ANTIPHOSPHOLIPID SYNDROME
Cellular Activation
The procoagulant phenotype in APS results from the synergy between aPL-mediated cellular activation of platelets, monocytes, plasmacytoid dendritic cells and ECs, as well as the disruption of natural anticoagulant and fibrinolytic systems (Fig. 1)
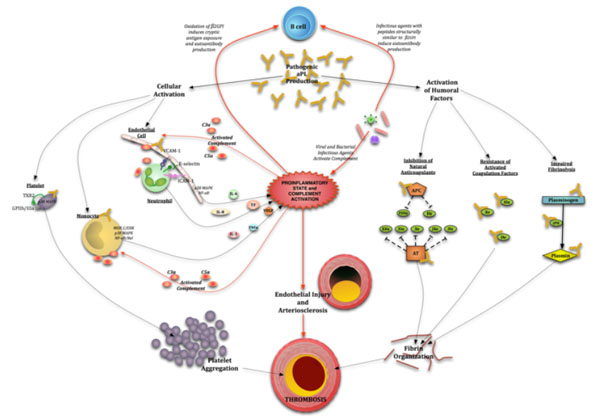
Pathogenetic mechanisms leading to thrombosis in antiphospholipid syndrome. Pathogenic aPL activate endothelial cells via p38 mitogen activated protein kinase (p38MAPK) and nuclear factor κB (NFkB) to produce several proinflammatory cytokines and adhesion molecules, monocytes via MEK-1/ERK, p38MAPK and NFkB/Rel to also produce proinflammatory cytokines, most importantly vascular endothelial growth factor (VEGF) and tissue factor (TF) and platelets via p38MAPK and NFkB to upregulate GPIIb/IIIa to induce platelet aggregation. Pathogenic aPL also bind several coagulation factors and their regulators: activated protein C (APC) and antithrombin (AT) to prevent inactivation of activated coagulation factors. Fibrinolysis is also inhibited by inactivation of tissue plasminogen activator (tPA) and plasmin. Infection/inflammation plays a central role in upregulating autoantibody production, causing endothelial injury and activating monocytes and neutrophils via complement-complement receptor interactions.
Platelet activation in APS occurs through the β2GPI-dependent interaction of aPL with phosphatidylserine, apolipoprotein E receptor 2’ (ApoER2’) and the GPIb( subunit of the GPIb-V-IX receptor on the platelet membrane [9-2]. This leads to the activation of the p38 mitogen activated protein kinase (p38MAPK) and subsequent phosphorylation of cytosolic phospholipase A2 (cPLA2) to produce thromboxane B2 (TXB2) and enhanced expression of GPIIb/IIIa, a major fibrinogen receptor. The ERK-1 (p44 MAPK), ERK-2 (p42 MAPK) pathways and other MAPK pathways, have a potential secondary role after initial activation through the p38 MAPK pathway [11]. The enhanced platelet expression of GPIIb/IIIa in turn leads to platelet aggregation, an important component in thrombus formation. A recent study has demonstrated a putative role for platelet factor 4 (PF4), a CXC chemokine secreted and bound by platelets, in the stabilization of dimeric β2GPI and subsequent binding to ant-β2GPI Abs and exposed phospholipids and receptors on the platelet surface [13].
Several monocyte and EC surface receptors bind β2GPI, providing suitable epitopes for aPL binding to these cells. Annexin A2 (AnnA2), present on both ECs and monocytes, binds anti-β2GPI/β2GPI complexes resulting in intracellular signaling cascade and release of pro-inflammatory cytokines [14]. However, AnnA2 on the cell surface lacks an intracellular tail, suggesting that a co-receptor or co-receptors would be required for intracellular signal transduction and subsequent cell activation [15]. Other studies confirmed that toll-like 4 (TLR4) acts as a co-receptor for annexin A2 binding to anti-β2GPI/β2GPI complexes and subsequent EC [16, 17] and monocyte [18] activation. Allen et al. have confirmed an essential role for TLR4 in EC activation by aPL, but mediated through the assembly of a multiprotein-signaling complex on the EC surface that includes AnnA2, TLR4, calreticulin and nucleolin [19]. The activation of endogenous TLR receptors (TLR7 and TLR8) by aPL in plasmacytoid dendritic cells and monocytes has also been shown to be a potentially important mechanism of initiation and/or propagation of the disease process in APS patients [20]. ApoER2’ is also expressed on ECs and in vitro and in vivo studies demonstrate a putative role for this receptor in aPL-mediated EC activation [21, 22].
APL-mediated activation of ECs by aPL results in p38 MAPK activation and nuclear factor κB (NF-κB) upregulation [23], while activation of monocytes results in phosphorylation of MEK-1/ERK proteins, p38 MAPK-dependent nuclear translocation and activation of NF-kB/Rel proteins [24]. EC and monocyte activation results in the increased expression of several pro-inflammatory and pro-coagulant factors including tissue factor (TF), vascular endothelial growth factor (VEGF), Flt-1 tyrosine kinase receptor, interleukin (IL)-6, IL-8, vascular cell adhesion molecule (VCAM)-1, intracellular adhesion molecule (ICAM)-1 and E-selectin [23-27]. These cytokines induce innate immune cell activation, stimulate the coagulation pathway and allow for leukocyte adhesion and thrombus formation in the presence of an inciting factor.
The complement system is an integral part of innate immunity, consisting of a number of small proteins including proteases, which catalyze a sequential series of reactions ultimately resulting in an amplification of innate immune responses. Studies done by Pierangeli et al, utilizing APS murine models of thrombosis, have highlighted the importance of complement C3, C5a/C5a-receptor interactions, C6 and the membrane attack complex (MAC) in aPL-mediated thrombosis [28-30]. Complement activation, as a result of the presence of inciting pro-inflammatory factors such as infection, and the subsequent generation of potent vasoactive mediators, likely occurs as a necessary intermediary step between cellular activation by aPL and thrombotic events characteristic of APS.
Coagulation and Fibrinolytic Systems
Antibodies to several coagulation factors and natural anticoagulants have been reported in APS patients, which act to prevent inactivation of activated coagulation factors and hinder the activity of anticoagulants. Anti-prothrombin antibodies (aPT) have been isolated from APS patients and these antibodies, particularly aPT complexed to PS (aPT/PS), have displayed LA activity, can induce TF expression and have thrombogenic properties in vivo [31-33]. Several aPT also display thrombin-binding activity and in their interaction with thrombin, prevent its inactivation by antithrombin (AT), promoting thrombosis by unchecked fibrinogen and platelet activation by thrombin [34, 35]. The presence of aPL with binding activity for activated factor IX (FIXa) and FXa in APS patients has also been reported, and several of these antibodies prevent the inactivation of FIXa and FXa by antithrombin [36, 37].
Antibodies to activated protein C (APC), a natural anticoagulant that functions by binding and inactivating FVa and FVIIIa, were reported as early as 1989 and displayed inhibitory activity, confirmed in subsequent studies [38, 39]. Antithrombin (AT) is another natural anticoagulant that performs its function by inactivating FXIIa, XIa, FXa, FIXa, FVIIa and thrombin, these actions being potentiated by heparin. The inhibition of heparin dependent activation of AT activity in vitro by up to 80% by isolated aPL has been described [40]. The inhibition of APC and AT activity potentiates a thrombogenic state since many activated factors remain unchecked.
A small pilot analysis of plasma samples from 25 APS patients showed that seven (28%) of these patients had IgG anti-plasmin antibodies [41]. Isolated aPL antibodies with activity against plasmin as well as whole IgG from APS patients, have been found to impair plasmin-mediated fibrinolysis [41, 42]. A subsequent study of the reactivity of aPL with tissue plasminogen activator (tPA), another fibrinolytic factor sharing homology with plasmin, showed that several of these aPL were able to reduce tPA activity in converting plasminogen to plasmin [43, 44]. Elevated plasminogen activator inhibitor type 1 (PAI-1) levels and decreased tPA release after venous occlusion in APS patients has also been reported [45].
FEATURES OF APS NEPHROPATHY
Histopathological Features
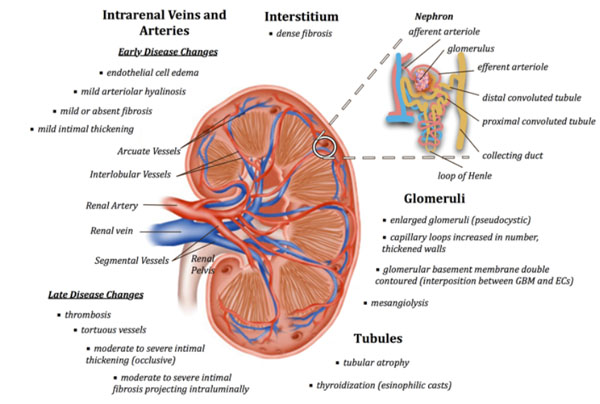
Thrombotic microangiopathy (TMA) lesions are considered acute lesions of APS nephropathy and chronic lesions include arteriosclerosis, fibrous intimal hyperplasia (FIH), fibrous obliteration of arteries and arterioles, and also focal cortical atrophy (FCA) (Fig. 2). These characteristic lesions have been described with increased frequency in primary APS patients as well as SLE patients positive for aPL, including secondary APS patients and non-APS lupus patients with aPL, compared to patients without aPL.
Mechanisms of thrombosis and possible targets. Numbers indicates the sites of action of the following drugs: 1 N-acetilcystein, 2 statins, 3 hydroxychloroquine, fluvastatin and FXa inhibitors(anticoagulants e.g. low molecular weight heparin, fondaparinux, rivaroxaban and edoxaban), 4 PDI inhibitors and hydroxychloroquine, 5hydroxychloroquine, 6 TLR7 inhibitors, 7 heparin, eculizumab, 8 belimumab
Thrombotic Microangiopathy
Acute lesions (TMA) represent the most characteristic lesion in APS nephropathy. It is defined by the presence of fibrin thrombi in the intra-renal vasculature tree and glomerular capillaries in the absence of inflammatory cells or immune deposits [46, 47]. Preglomerular arterioles, small interlobular arteries and glomerular capillaries are most often affected [46]. During this acute phase, EC swelling occurs, which leads to narrowing or occlusion of the vessel lumen. This initial narrowing is further aggravated by fibrin thrombi containing fragmented erythrocytes, leucocytes and eosinophilic fibrinoid material [46, 48]. Glomeruli can also show associated mesangiolysis and diffuse mesangial interposition with numerous double contours [49].
Arteriosclerosis, Fibrous Intimal Hyperplasia and Thrombus Organization
Arteriosclerosis lesions consist of fibrous intimal thickening with reduction of affected arteries, associated with arteriolar hyaline and arteriolosclerosis [46, 48, 50]. The intima of these affected vessels tend to be much more cellular than what is normally seen in typical arteriosclerosis lesions of aging. Fibrous intimal hyperplasia (FIH) is characterized by tortuous intra-renal arteries, with thickened intima due to an intense myofibroblastic intimal cellular hyperplasia (α and γ smooth cells) and mucoid extracellular matrix [46]. The media is either proliferative, containing hypertrophic myocytes, or atrophic and fibrous. Subsequent thrombus organization, often seen in patients with extensive FIH, results in complete vascular occlusion, which may be re-cannalized through endothelialization of existing thrombi to restore blood flow. Repeated thrombosis/recanalization ultimately leads to the development of fibrotic intimal lesions that project into vessel lumen, resulting in partial or complete occlusion and eventually glomerular basement changes [46, 48].
Focal Cortical Atrophy
Focal cortical atrophy is characterized by dense interstitial fibrosis, massive tubular atrophy and tubular thyroidization, ie- atrophic tubules containing eosinophilic casts [46, 50]. It most likely occurs as a function of numerous occlusions in vessels supplying the renal cortex with subsequent ischemic infarction [46]. These triangular lesions extend from the superficial cortex below the renal capsule to the deep cortex, are irregularly distributed and retract from the surrounding normal cortex separated by sharp borders. They are typically associated with the vascular lesions of fibrous intimal hyperplasia (FIN) with recanalizing thrombi and arteriolar occlusion [46]. Glomeruli in FCA lesions are often small and sclerotic, five or six globally sclerosed glomeruli clustered together. Occasionally these groups of glomeruli are voluminous rather than being sclerosed.
Clinical Features
Clinically, the most common and earliest presenting feature of APS nephropathy is systemic hypertension. Nochy et al. evaluated 16 primary APS patients with renal involvement and reported that hypertension was present in 93% of the patients studied, the severity ranging from mild to severe and even malignant [46]. In this study, malignant hypertension was present in only one patient who also developed microangiopathic hemolytic anemia and end-stage renal disease requiring dialysis. Hypertension was the only presenting feature of APS nephropathy in several cases but was variably associated with renal insufficiency, proteinuria or hematuria. In a study of APS nephropathy in systemic lupus erythematosus (SLE) patients, hypertension was a presenting feature in 77.1% of SLE patients with APS nephropathy and was the only clinical feature that remained significantly associated with APS nephropathy after multivariate regression analysis [50]. Other studies of renal disease in SLE patients with aPL report prevalence rates of hypertension approaching 100% [51]. The typical thrombotic microangiopathic intrarenal lesions that characterize APS nephropathy generate systemic hypertension, which in turn may worsen these lesions leading to progressive damage. The appearance of hypertension in a patient with primary or secondary APS could indicate the development of nephropathy associated with this disorder or stenosis or occlusion of the renal artery [46, 50-52]. As such, APS patients who develop hypertension, even in the absence of proteinuria, hematuria or decreased glomerular filtration rate, must be extensively investigated, including renal biopsy [53].
Other common clinical features, occurring most often with accompanying hypertension, are acute or chronic renal insufficiency, proteinuria and intermittent hematuria [46]. In primary APS patients, renal insufficiency has been reported in up to 87% cases in one study, 75% with chronic insufficiency [46]. Insufficiency was mild in most cases, serum creatinine levels at the time of renal biopsy ranging from 1.02 to 2.50 mg/dl (mean 1.64 mg/dl), but one patient subsequently progressed to end stage renal failure. Proteinuria occurred in 75% of cases, being generally mild (range 0.2 - 3.0 g/day), however nephrotic range proteinuria occurred in one patient. Hematuria occurred in 56% of patients, while microangiopathic hemolytic anemia occurred rarely. Other studies of APS nephropathy in primary APS patients also report the high prevalence of hypertension, mild insufficiency, mild proteinuria and intermittent hematuria, with the rare occurrence of nephrotic range proteinuria and end-stage renal disease [47, 51-55].
In a large study of SLE patients, APS nephropathy was present in 39.5% of SLE patients positive for aPL [50]. APS nephropathy was present in 67% of secondary APS patients but in only 31.7% of non-APS lupus patients with aPL and this was similar to prevalence rates reported in earlier studies [48, 50]. SLE patients with APS nephropathy have been reported in several studies to have increased risk for the development of hypertension, raised serum creatinine levels, and progression of histologic lesions independent of the effect of lupus nephritis and all were associated with a poor renal outcome [48, 50, 55, 56]. However, the prevalence of chronic renal insufficiency, proteinuria and nephrotic syndrome in SLE patients with APS nephropathy has varied across studies [55-57].
The clinical evaluation of any APS patient who has renal pathology, should include a thorough assessment of other possible cause of thrombotic microangiopathy (TMA), because the characteristic clinical findings for APS nephropathy are not disease specific [53, 58]. Patients with TTP and HUS are much more likely to present with severe thrombocytopenia and microangiopathic hemolytic anemia, which are relatively uncommon in APS nephropathy [59]. In contrast, the presence of aPL or clinical and laboratory features typical of CAPS are strong indicators of APS nephropathy. Ultimately, the diagnosis is made upon finding classical histo-pathological lesions on renal biopsy, and even so, other causes of TMA need to be excluded.
ASSOCIATION OF APS NEPHROPATHY AND ANTIPHOSPHOLIPID ANTIBODIES
Several studies have established a link between the presence of aPL and the occurrence of APS nephropathy. APS nephropathy has been detected in primary and secondary APS patients (Table 1) and non-APS lupus patients positive for aPL (Table 2) at significantly higher rates than in patients with SLE who were negative for aPL [48, 50, 60-68]. This indicates that the characteristic lesions seen in APS nephropathy occur as a result of the pathological action of aPL. Interestingly, APS nephropathy occurs with significantly higher frequency in primary and secondary APS patients, compared to non-APS lupus patients with aPL positive for aPL [7, 50]. The prevalence of APS nephropathy in primary APS patients with renal disease was almost always 100% across several studies [46, 47, 55, 69-72], while the prevalence in secondary APS patients with SLE and biopsy-proven renal involvement ranged from 67% to 100% across studies [48, 50, 51, 70, 73-75]. The association of APS nephropathy with aPL, seems to be strongest for LA, but has been reported for anticardiolipin antibodies as well. The characteristic acute and chronic lesions of APS nephropathy have been detected in all subgroups of patients with positive aPL, however, acute TMA lesions seemed to be more common in patients with CAPS while there was no apparent difference in the relative frequency of chronic lesions among these subgroups [55].
Prevalence of APS nephropathy in primary and secondary APS patients compared to SLE patients without antiphospholipid antibodies.
| Study | Comparison | APS Nephropathy* | p-Value |
|---|---|---|---|
| Moss, Isenberg, 2001 [66] | 14 PAPS/SAPS pts 72 SLE/non-APS pts neg for aPL |
5/14 (35.7%) 0/72 (0%) |
0.0002 |
| Daugas et al., 2002 [54] | 76 SAPS/SLE pts w. renal Dx pos for aPL 15 SLE pts w. renal Dx neg for aPL |
28/76 (36.8%) 3/15 (20.0%) |
0.063 |
| 33 SAPS/SLE pts w. renal Dx pos for LA 77 SAPS/SLE pts w. renal Dx neg for LA |
20/33 (60.6%) 16/77 (20.8%) |
0.0001 | |
| 65 SAPS/SLE pts w. renal Dx pos for aCL 25 SAPS/SLE pts w. renal Dx neg for aCL |
21/65 (32.3%) 8/25 (32.0%) |
0.98 | |
| Tektonidou et al., 2004 [47] | 81 SAPS/SLE pts w. renal Dx pos for aPL 70 SLE pts w. renal Dx neg for aPL |
32/81 (39.5%) 3/70 (4.3%) |
< 0.01 |
| 20 SAPS/SLE pts w. renal Dx pos for LA 131 SAPS/SLE pts w. renal Dx neg for LA |
16/20 (80.0%) 19/131 (14.5%) |
< 0.01 | |
| 81 SAPS/SLE pts w. renal Dx pos for aCL 70 SAPS/SLE pts w. renal Dx neg for aCL |
30/81 (37.0%) 5/70 (7.1%) |
< 0.01 | |
| Salvario et al., 2011 [67] | 39 SAPS/SLE pts w. renal Dx pos for aPL 34 SLE pts w. renal Dx neg for aPL |
9/39 (23.1%) 0/34 (0%) |
0.003 |
| 22 SAPS/SLE pts w. renal Dx pos for LA 51 SAPS/SLE pts w. renal Dx neg for LA |
7/22 (31.8%) 2/51 (3.9%) |
0.002 | |
| 25 SAPS/SLE pts w. renal Dx pos for aCL 48 SAPS/SLE pts w. renal Dx neg for aCL |
7/25 (28.0%) 2/48 (4.2%) |
0.006 | |
| Erre et al., 2014 [68] | 7 SAPS pts w. LN 41 SLE/non-APS pts w. LN |
4/7 (57.1%) 8/41 (19.5%) |
0.05 |
| 23 SAPS/SLE pts w. LN pos for aPL 25 SLE pts w. LN neg for aPL |
8/23 (34.7%) 4/25 (16.0%) |
NS | |
| 11 SAPS/SLE pts w. LN pos for LA 37 SAPS/SLE pts w. LN neg for LA |
6/11 (54.5%) 6/37 (16.2%) |
0.02 | |
| 22 SAPS/SLE pts w. LN pos for aCL 26 SAPS/SLE pts w. LN neg for aCL |
8/22 (36.3%) 4/26 (15.4%) |
NS | |
| 10 SAPS/SLE pts w. LN pos for aCL and LA 38 SLE pts w. LN not aCL and LA pos |
6/10 (60.0%) 6/38 (15.8%) |
0.01 |
* APS Nephropathy defined as presence of thrombotic microangiopathy in early studies and all characteristic acute and chronic lesions in later studies. ¶aPL refers to all antiphospholipid antibodies, for most studies including aCL and LA. aCL- anticardiolipin, APS - antiphospholipid syndrome, Dx - disease, LA- lupus anticoagulant, LN - lupus nephritis, NS - not significant, PAPS - primary APS, pts - patients, SAPS - secondary APS, SLE - systemic lupus erythematosus, w. – with.
Prevalence of APS nephropathy in non-APS lupus patients positive for antiphospholipid antibodies compared to SLE patients without aPL.
| Study | Comparison | APS Nephropathy* | p-Value |
|---|---|---|---|
| Kant et al., 1981 [60] | 9 SLE pts pos for LA 96 SLE pts neg for LA | 7/9 (77.8%) 27/96 (28%) | < 0.001 |
| Glueck et al., 1985 [61] | 18 SLE pts pos for LA 67 SLE pts neg for LA | 14/18 (77.8%) 25/67 (37.3%) | < 0.005 |
| Farrugia et al., 1992 [62] | 33 SLE pts w. renal Dx pos for LA 32 SLE pts w. renal disease neg for LA | 5/33 (15.2%) 0/32 (0%) | 0.05 |
| Miranda et al., 1994 [65] | 9 SLE pts w. renal Dx pos for aCL 27 SLE pts w. renal Dx neg for aCL | 3/9 (33.3%) 5/27 (18.5%) | 0.38 |
| Bhandari et al., 1998 [63] | 24 SLE pts w. LN pos for aCL 26 SLE pts w. LN neg for aCL | 17/24 (70.8%) 6/26 (23.1%) | 0.0015 |
| Naiker et al., 2000 [64] | 18 SLE pts w. renal Dx pos for aCL 22 SLE pts w. renal Dx neg for aCL | 5/8 (27.8%) 2/22 (9.1%) | 0.025 |
* APS Nephropathy defined as presence of thrombotic microangiopathy in early studies and all characteristic acute and chronic lesions in later studies. aCL- anticardiolipin, aPL - antiphospholipid antibodies, APS - antiphospholipid syndrome, Dx - disease, LA- lupus anticoagulant, LN - lupus nephritis, PAPS - primary APS, pts - patients, SAPS - secondary APS, SLE - systemic lupus erythematosus, w. - with.
At the 13th International Congress on Antiphospholipid Antibodies, held in Galveston, Texas in April 2010, a task force comprising experts in the field was created to examine the association of aPL with several non-criteria manifestations of APS, including APS nephropathy. They concluded that there was level II-III evidence that aPL correlates with both acute and chronic lesions of APS nephropathy [7]. This Task Force also evaluated the accuracy of APS nephropathy for the detection of patients with APS, comparing primary and secondary APS patients with patients negative for aPL. They reported that the sensitivity of APS nephropathy for predicting APS was only 64%, while the specificity, positive predictive value and negative predictive value were much higher at 96%, 85% and 87% respectively.
CONCLUSION
Renal involvement is a well-recognized clinical manifestation of APS, and APS nephropathy is seen as the characteristic clinico-morphological manifestation of renal disease in APS. The pathogenesis of APS nephropathy seems to be the occurrence of thrombotic lesions in the intra-renal vasculature, leading to thrombotic microangiopathy and over time the development of chronic vaso-occlusive lesions, fibrosis and atrophy. Recent years have seen an improved characterization of these lesions and a clear link between the presence of aPL and occurrence of APS nephropathy has been established. What seems clear is that the occurrence of hypertension, proteinuria, hematuria or renal insufficiency in patients with APS or SLE patients positive for aPL, is enough to warrant thorough investigation for APS nephropathy, including renal biopsy. The strong association of aPL positivity with the occurrence of APS nephropathy and the specificity of this manifestation for predicting APS disease, suggest that APS nephropathy should be included in APS classification criteria.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

