All published articles of this journal are available on ScienceDirect.

Genetic Diseases of the Kidney
Abstract
The number of genes associated with renal disease is increasing every day and this has led to a clearer understanding of the pathophysiology of renal disease in many disorders. It is also appreciated now that a genetic mutation(s) underlie many renal syndromes. Genetic testing may also offer the possibility to diagnose some renal diseases without the need for a renal biopsy. It also allows the prenatal diagnosis of certain renal diseases in at risk fetuses or identification of potential renal disease before it has become manifest. Finally, identification of a specific gene mutation holds the possibility of correction though gene therapy in the future. It is increasingly clear that many renal disorders in pediatrics are a consequence of genetic mutations. In the future, genetic testing will become as easy and as common as ordering a serum creatinine today.
1. INTRODUCTION
The number of genes associated with renal disease is increasing every day and this has led to a clearer understanding of the pathophysiology of renal disease in many disorders. It is also appreciated now that a genetic mutation(s) underlie many renal syndromes. Genetic testing also offers the possibility to diagnose some renal diseases without the need for a renal biopsy. It also allows the prenatal diagnosis of certain renal diseases in at risk fetuses or identification of potential renal disease before it has become manifest. Finally, identification of a specific gene mutation holds the possibility of correction though gene therapy, although gene therapy remains only a promise at this point in time.
The first step in identifying a genetic renal disorder is to consider it. A gene mutation should be thought of if another family member is affected, if there are other malformations in addition to the renal disease, or if the clinical course is different from that of the typical patient with this disorder.
However, there are a number of problems associated with genetic testing. First, the identification of a gene mutation for many renal diseases remains a research procedure that requires informed consent and often this information cannot be used for making clinical decisions. Further, most genetic renal disorders are caused by a number of mutations in a given gene and can be caused by a mutation in more than one gene. This makes the identification of the precise mutation in the right gene in a given patient difficult. Commercially available genetic testing laboratories often only identify the common mutations, thus missing some patients. Not all insurance carriers will pay for genetictesting. Finally, genetic testing itself carries the risk of labeling someone as a carrier or a potential victim of a disease that may have long-term implications.
2. GENETIC DISORDERS AFFECTING THE GLOMERULUS
2.1. Podocyte - Nephrotic Syndrome (Table 1, Fig. 1)
Genetic disorders of the podocyte.
| Gene | Protein | Inheritance | Onset | Comments |
|---|---|---|---|---|
| NPHS1 | Nephrin | AR | Birth | Placentomegaly, prematurity, Finnish CNS |
| NPHS2 | Podocin | AR | 3mo-5y | |
| PLCE1 | PLCƐ1 | AR | Birth -1y | Mesangial sclerosis |
| PTPRO | PTPRO (GLEPP1) | AR | Childhood | |
| MYOIE | MOYIE | AR | 1-9y | Non-muscle myosin |
| ITGA3 | α1 and α2 integrin | AR | Infancy | Pulmonary and skin disease |
| WDR73 | WD40-repeat-containing protein | AR | Infancy to childhood | Galloway-Mowat syndrome |
| WT1 | WTI protein | AD | Infancy | Denis-Drash Syndrome -NS, Wilms tumor, male pseudohermaphroditism |
| WTI | WT1 protein | AD | Childhood | Frasier syndrome - NS, male pseudohermaphroditism, gonadoblastomas |
| ACTN4 | α-Actinin 4 | AD | Adult | |
| TRPC6 | TRPC6 | AD | Adult | |
| INF2 | INF2 | AD | Adult | |
| ARHGAP24 | Arhgap24 | AD | Adult |
Genetic disorders of the glomerular basement membrane disorders.
| Gene | Protein | Inheritance | Onset | Comments |
|---|---|---|---|---|
| α-5 chain | α-5 chain | X-linked | Early childhood | Alport syndrome - ESRD at 2-4 decade, Deafness |
| Heterozygous α-3 or α-4 mutation | α-3 or α-4 chain | AD | Early childhood | Thine basement membrane disease -Familial hematuria |
| Homozygous α-3 or α-4 mutation | α-3 or α-4 | AR | Early childhood | Alport syndrome - ESRD at adolescence, deafness |
| LAMB2 | Laminin 2 | AR | Infancy | Pierson syndrome - Eye disease, MR |
| LMX1B | LMX1B | AD | Variable | Nail-Patella syndrome - Dysplastic nails and patellae, variable degree of renal involvement |
| ITGA3 | Integrin α3 subunit | AR | Infancy | Kidney disease, lung disease, epidermolysis bullosa |
Lipid and mitochondrial abnormalities causing NS.
| Gene | Protein | Inheritance | Onset | Comments |
|---|---|---|---|---|
| ApoE | Apolipoprotein E | AD | Adulthood | Lipid glomerulopathy with lipid thrombi, Asian descent |
| LCAT | Lecithin:cholesterol acyltransferase | AR | Adulthood | Corneal opacities, hepatosplenomegaly, anemia, CKD |
| CoQ6 | Coenzyme Q6 | AR | Infancy | NS, sensorineural deafness, ESRD |
Proximal tubule genetic disorders.
| Gene | Protein | Inheritance | Onset | Comments |
|---|---|---|---|---|
| SLC5A2 | SGLT2 | AD | Birth | Benign glucosuria |
| SLC3A1 | rBAT | AR | Infancy to adulthood | Cystinuria, dibasic aminoaciduria, renal stones |
| SLC7A9 | B0,+AT1 | AR | Infancy to adulthood | Cystinuria, dibasic aminoaciduria, renal stones |
| SLC34A3 | Na-PO4 transporter | AR | Infancy | Hereditary hypophosphatemic rickets |
| CAII | Carbonic anhydrase II | AR | Infancy | FTT, proximal and distal RTA, osteopetrosis |
| SLC4A4 | NBC1 | AR | Infancy | FTT, proximal RTA, developmental delay, eye disease |
| CTNS | Cystinosin | AR | Infancy | Cystinosis -Fanconi syndrome, FTT, hypothyroidism, eye disease, ESRD |
| CLCN5 | CLC-5 chloride channel | AR | Infancy | Dent disease - kidney stones, nephrocalcinosis, Fanconi syndrome, ESRD |
| OCRL | INPP5F | X-Linked | Infancy | Dent disease |
| OCRL | INPP5F | X-Linked | Infancy | Lowe syndrome - developmental delay, ocular disease, Fanconi syndrome, ESRD |
| SLC22A12 | URAT1 | AR | Infancy | Hereditary hypouricemia |
Ascending limb genetic disorders.
| Gene | Protein | Inheritance | Onset | Comments |
|---|---|---|---|---|
| SLC12A2 | NKCC2 | AR | Infancy | Bartter syndrome type 1 - hypokalemia, metabolic alkalosis, hypercalciuria, FTT |
| KCNJ1 | ROMK | AR | Infancy | Bartter syndrome type 2 |
| CLCNKB | CLCNKB | AR | Infancy | Bartter syndrome type 3 |
| BSND | Bartin | AR | Infancy | Bartter syndrome type 4, deafness |
| CASR | Calcium-sensing receptor | AD | Childhood to adulthood | Bartter type 5, mild hypokalemia with hypocalcemia |
| CLDN-16 | Claudin-16 | AR | Childhood | FHHN - familial, hypomagnesemia, hypercalciuria, nephrocalcinosis, ESRD |
| CLDN-19 | Caludin-19 | AR | Childhood | FHHN |
| UMOD | Uromodulin | AD | Adolescence | Hyperuricemia, gout, ESRD |
Genetic disorders of the distal convoluted tubule and collecting duct.
| Gene | Protein | Inheritance | Onset | Comments |
|---|---|---|---|---|
| SLC12A3 | NCCT | AR | Childhood to adulthood | Gitelman syndrome - hypokalemia, hypomagnesemia, tetany |
| SNN1B | Β subunit of ENaC | AD | Childhood | Liddle syndrome - hypertension, hypokalemia, alkalosis, suppressed aldosterone and renin |
| SNN1G | Γ subunit of ENaC | AD | Childhood | Liddle syndrome |
| HSD11B2 | 11β-hydroxysteroid dehydrogenase | AR | Childhood | Apparent mineralocorticoid excess - Hypertension, suppressed aldosterone and renin |
| WNK1 | WNK1 protein | AR | Childhood | Gordon syndrome - hypertension, hyperkalemia, metabolic acidosis |
| WNK4 | WNK4 protein | AR | Childhood | Gordon syndrome |
| MLR | Mineralocorticoid receptor | AD | Childhood | AD pseudohypoaldosteronism type1 - hyponatremia, hyperkalemia, salt wasting |
| SNN1A | α-subunit of ENaC | AR | Infancy | AR pseudohypoaldosteronism type 1 - severe salt wasting, hyperkalemia, hyponatremia |
| SNN1B | β-subunit of ENaC | AR | Infancy | AR pseudoaldosteronism |
| SNN1G | γ-subunit of ENaC | AR | Infancy | AR pseudoaldosteronism |
| AE1 | AE1 | AD | Infancy | dRTA |
| ATP6V1B1 | β-subunit of H+-ATPase | AR | Infancy | dRTA, deafness |
| ATP6V0A4 | α-subunit of H+-ATPase | AR | Infancy | dRTA |
| AVPR2 | AVPreceptor | X-linked | Infancy | Nephrogenic diabetes insipidus |
| AQP2 | Aquaporin 2 | AR | Infancy | Nephrogenic diabetes insipidus |
Cystic kidney disease.
| Gene | Protein | Inheritance | Onset | Comments |
|---|---|---|---|---|
| PKD1 | Polycystin 1 | AD | Childhood to adulthood | Large renal cysts, liver cysts |
| PKD2 | Poycystin 2 | AD | Childhood to adulthood | Large renal cyst, liver cysts |
| PKHD | Fibrocystin | AR | Infancy to childhood | Nephromegaly with microcysts, hepatic fibrosis, ESRD |
| NPHP1 | Nephrocystin-1 | AR | Childhood | Nephronophthisis(NPHP) type 1 - interstitial fibrosis with microcysts |
| Inversin | Inversin | AR | Infancy to childhood | NPHP type 2 -early onset, situs in versus |
| NPHP3 | NPHP3 | AR | Adolescence | NPHP type 3 |
| NPHP4 | Nephroretinin | AR | Adolescence | NPHP type 4 |
| NPHP5/IQCB1 | NPHP5/IQ motif containing B1 | AR | Childhood | NPHP type 5 |
| NPHP6/CEP2901 | NPHP6/centrosome protein Cep290 | AR | Childhood | NPHP6 |
| NPHP7/GLIS2 | GLIS family zinc finger protein 2 | AR | Adolescence | NPHP7 |
| NPHP8/RPGRIP1L | RPGRIP1L | AR | Childhood | NPHP8, Meckel syndrome, Joubert syndrome |
| NPH9/NEK8 | NIMA-related kinase | AR | Childhood | NPHP9 |
| SDCCAG8 | SDCCAG8 | AR | Childhood | NPHP10, Senior-Loken syndrome |
| TMEM67 | Transmembrane protein 67 | AR | Childhood | NPHP11, hepatic fibrosis |
| TTC21B | Tetratricopeptide repeat domain-containing protein 21B |
AR | Childhood | NPHP12 |
| WDR19 | WD repeat-containing protein 19 | AR | Childhood | NPHP13 |
The majority of infants, especially in the first 3 months of life, with nephrotic syndrome have a mutation in both copies of one of several different genes [1]. The first to be described was congenital nephrotic syndrome of the Finnish type that has an incidence in Finland of 1 in 10,000 newborns [2]. It is much rarer in other countries. It is caused by a defect in the gene, NPHS1, that codes for the protein, nephrin, found in the slit diaphragm. Infants with Finnish congenital nephrotic syndrome have massive proteinuria that begins in utero and are born prematurely with very large placentas. Edema is either present at birth or appears shortly afterwards and is difficult to manage. There is no specific therapy except nephrectomy and ultimately renal transplantation.
Other causes of congenital nephrotic syndrome include mutations in NPHS2, which codes for podosin [3], PLCE1 [4], which codes for PLCƐ1, and WT1 [5] which codes for Wilms tumor suppressor gene. Podosin is important in the formation of the slit diaphragm and intracellular signaling. PLCƐ1 or phospholipase C epsilon generates diacylgycerol and inositol 1,4,5-triphosphate that is important for intracellular signaling in the podocyte. A mutation in both copies of either gene leads to the nephrotic syndrome and rapid progression to end-stage renal disease (ESRD). Infants with genetic mutation mediated nephrotic syndrome are resistant to steroid therapy, although a few infants with a PLCE1 mutation have responded to cyclosporine [5].
Denys-Drash syndrome [5], due to a mutation in WT1, presents in infancy with nephrotic syndrome and in males with ambiguous to female genitalia. These patients have diffuse mesangial glomerulosclerosis on kidney biopsy and they also develop Wilms tumor. The nephrotic syndrome is refractory to immunosuppressive therapy and patients with it usually progress to renal failure by age 3 years. Galloway-Mowat syndrome is a rare autosomal recessive condition that is characterized by microcephaly, neurologic impairment and nephrotic syndrome [6]. It is probably genetically heterogeneous, but a mutation in WDR73 was described in a single family. WDR73 codes for a WD40-repeat-containing protein of unknown function, but appear to play a role in maintenance of cell architecture and survival.
While the majority of pediatric patients with nephrotic syndrome respond to steroids, approximately 20-40% do not. Of those that do not, it is estimated that as high as a third have a genetic disorder causing the nephrotic syndrome [7]. The most common cause of this is a mutation in NPHS2 [8, 9]. A few children with steroid-resistant nephrotic syndrome have a mutation in PTPRO or MYO1E [10]. The patho-genesis of nephrotic syndrome in these disorders is unclear, but both cause foot process effacement, focal segmental glomerulosclerosis [FSGS], and rapid progression to ESRD. Frasier syndrome [11]is caused by a different mutation in WTI from that which causes the Denys-Drash syndrome and causes male pseudohermaphroditism and nephrotic syndrome in late childhood with FSGS [12]. Affected individuals can also develop gonadoblastomas.
In contrast to genetic causes of nephrotic syndrome in infancy and childhood that are mainly inherited as autosomal recessive traits, genetic causes of nephrotic syndrome that present in adolescence and adulthood are usually inherited as an autosomal dominant trait and with the pathological changes typical of FSGS. The first to be described was a mutation in the gene coding for (-actinin-4 protein [13]. (lpha-actinin-4 plays an important role in maintaining the cytoskeleton of the podocyte. A mutation in the gene coding for the transient receptor potential cation channel 6 TRPC6 protein [14] also causes autosomal dominant nephrotic syndrome. Abnormalities in channel activity leads to increased intracellular calcium in the podocyte that interferes with cell function and increased apoptosis of the podocyte. Mutations in the formin gene, INF2, cause autosomal dominant FSGS [15]. INF2 protein is involved in the regulation of acting and maintenance of the cytoskeleton of the podocyte. Similarly, a mutation in the gene ARGHGAP24 coding for the Rac1 regulatory protein, Arhgap24, causes dysregulation of the podocyte actin cytoskeleton and autosomal dominant FSGS [16].
2.2. Basement Membrane Disorders (Table 2)
Hereditary nephritis or Alport syndrome is caused by a mutation in the genes coding for the alpha-3,4, or 5 chains of type IV collagen [17]and causes progressive glomerular disease often associated with neural deafness and eye abnormalities. It is the most common basement membrane disorder causing kidney disease with an incidence of 1/5000 live births and accounts for 2% of the new cases of ESRD [18]. The initial manifestation is microscopic hematuria that occurs early in life with progressive renal disease that leads to ESRD by the second to third decade of life. The most common form of Alport syndrome is a mutation in COL4A5 gene located on the X chromosome coding for the alpha-5(IV chain and accounts for about 80% of Alport patients. Males are severely affected while most females have only hematuria, but some female carriers do develop renal failure. Approximately 15% of Alport patients have an autosomal recessive inheritance pattern and the mutation is in either the COL4A3 or COL4A4 genes that code for the alpha-3 and alpha-4 chains of type IV collagen, respectively [18].
Thin basement membrane disease is a relatively common familial disorder and is characterized by a glomerular basement membrane that is thinner than normal [19]. Patients affected by this disorder have isolated hematuria that is inherited in an autosomal dominant pattern. The defect is a mutation in one of the COL4A3 or COL4A4 genes [17-19]. These are the same genes associated with autosomal recessive Alport syndrome. Patient with thin basement disease rarely have significant proteinuria or progress to renal failure. They also don’t develop hearing loss or eye changes.
Nail-patella syndrome is a rare autosomal dominant condition characterized by hypoplastic or absent patellae, dysplastic nails and elbows, iliac horns, and proteinuria [20, 21]. Nail-patella syndrome is caused by a mutation in the transcription factor LMX1B. Proteinuria occurs in about half of affected individuals and a minority of these patients progress to renal failure. The glomerular basement membrane has a characteristic “moth-eaten” appearance from multiple irregular lucencies.
Pierson syndrome is a rare autosomal recessive disorder caused by a mutation in LAMB2 which codes for laminin β2 [22]. Renal disease usually starts early in life with nephrotic syndrome and progresses to ESRD within the first year of life. They also have buphthalmos, microcoria, hypotonia and profound psychomotor retardation. They have diffuse basement membrane abnormalities with mesangial sclerosis.
Mutations of ITGA3, which encodes for the transmembrane integrin α3 subunit, can cause congenital nephrotic syndrome. Infants with this mutation had marked irregularities of the basement membrane in the kidney, lung, and epidermis with interstitial lung disease and epidermolysis bullosa [23].
2.3. Lipid Abnormalities Causing Nephrotic Syndrome (Table 3)
Liproprotein glomerulopathy presents in adulthood with proteinuria and rapid progression to ESRD. It occurs mainly in individuals of Asian ethnicity. There is extensive deposition of laminated lipid thrombi in glomerular capillaries and affected individuals have type III hyperlipidemia [24]. Lecithin: cholesteraol acyltransferase deficiency is an autosomal recessive abnormality characterized by lipid abnormalities, corneal opacities, nephrotic syndrome and progressive renal disease. The basement membrane has a “vacuolized” appearance from lipid droplets [25].
2.4. Glomerular Mitochondrial Disorders (Table 3)
Mutations of the mitochondrial enzyme CoQ6 cause nephrotic syndrome and sensorineural deafness [26]. CoQ6 is necessary for the biosynthesis of CoQ10 which is essential for proper functioning of the electron transport chain. Nephrotic syndrome associated with CoQ6 mutations is inherited in an autosomal recessive pattern and all children progressed to ESRD by late childhood.
3. GENETIC DISORDERS AFFECTING THE PROXIMAL TUBULE (FIG. 2, TABLE 4)
Hereditary renal glucosuria is a rare disorder occurring in 1 in 20,000 individuals and is marked by glucosuria when the blood glucose in the normal range [27]. It is caused by a mutation in the gene, SLC5A2, that codes for the SGLT2 glucose transporter [28]. It is inherited as a co-dominant trait, thus individuals with a single gene mutation have glucosuria. Affected individuals are otherwise normal.
Cystinuria is characterized by the excessive excretion of cysteine and the dibasic amino acids ornithine, lysine and arginine [29, 30]. Cystine is relatively insoluble when its concentration exceeds 250mg/L and affected individuals have recurrent stones disease. It occurs with a incidence of 1/20,000 and is caused by a mutation in both SLC3A1 genes, which codes for the protein rBAT, or both SLC7A9 genes, which codes for the protein b0,+AT1 [30]. These two proteins form a heterodimer responsible for cystine and dibasic amino acid transport across the proximal tubule brushborder membrane. Patients that are compound heterozygotes also have cystinuria.
Hereditary hypophosphatemic rickets with hypercalciuria is a rare, autosomal recessive disorder cause of hypophosphatemia [31]. It is characterized by mutations in the SLC34A3 gene which codes for the Na-PO4 transporter in the proximal tubule brushborder. The hypophosphatemia causes excessive production of 1,25-vitamin D and hypercalciuria. This disorder is distinct from the more common X-linked hypophosphatemic rickets that is caused by abnormalities in PEX that regulates fibroblast growth factor 23 and phosphate reabsorption [32].
Inherited proximal renal tubular acidosis RTA can be caused by a mutation in both CA2 genes coding for carbonic anhydrase II [33]. Patients with this disorder also have distal RTA and osteopetrosis. Proximal RTA can also be caused by a mutation in both SLC4A4 genes, which encodes for NBC1 [34]. NBC1 transports both sodium and bicarbonate across the basolateral membrane. Affected children also have psychomotor retardation and ocular abnormalities.
Fanconi syndrome is a disorder characterized by a global dysfunction of the proximal tubule, leading to hypokalemia, hypophosphatemia, acidosis, glucosuria and aminoaciduria. Many inherited metabolic diseases can cause transient Fanconi syndrome, except for cystinosis [35], Lowe synd-rome [36, 37] and Dent disease [38]. Cystinosis is another rare autosomal recessive disease caused by a mutation in the CTNS gene, coding for cystinosin [39]. Cystinosin mediates the transport of cystine from the lysosome to the cytoplasm and affected individuals have excessive levels of cysteine in virtually all cells of the body [40]. Infant with cystinosis soon develop the Fanconi syndrome and later progressive renal dysfunction. With advent of cysteamine and renal transplantation to treat patients with cystinosis, numerous other organ dysfunctions have become evident including, hypothyroidism, diabetes mellitus, cardiovascular, ocular, and neuromuscular disorders [41].
Dent disease is an X-linked recessive disorder character-ized by low-molecular-weight proteinuria, hypercalciuria, phosphaturia, aminoaciduria, glucosuria, nephrolithiasis and nephrocalcinosis [35, 38]. Renal failure is common and may occur by late childhood. The disorder is caused by either a mutation in the CLCN5 gene [42] which codes for the CLC-5 chloride channel or in the OCRL gene [43] which codes for phosphotylinositol-4,5-biphosphate 5-phosphatase INPP5P. CLC-5 chloride channel activity is important for recycling megalin to the brushborder membrane [36]. Megalin plays an important role in solute reabsorption in the proximal tubule. INPP5P is involved in actin polymerization in the Golgi apparatus and defective functioning of this enzyme leads to either Lowe syndrome or Dent disease.
Lowe syndrome is another X-linked disorder causing Fanconi syndrome along with cataracts and other eye abnormalities and mental retardation [36-38]. It is caused by a mutation in the OCRL gene [44]. Lowe syndrome patients typically develop ESRD by the third to fourth decade of life.
Hereditary renal hypouricemia is a rare autosomal recessive disorder characterized by hypouricemia and increased uric acid clearance [45]. It is caused by a mutation in both SLC22A12 genes which code for the brushborder membrane protein URAT1. Most individuals with renal hypouricemia are asymptomatic, although a few affected individuals have had exercise-induced acute kidney injury.
4. ASCENDING LIMB DISORDERS (FIG. 3, TABLE 5)
The most common ascending limb disorder is Bartter syndrome which is an autosomal recessive disease caused by a number of genetic mutations [46]. Bartter syndrome is characterized by hypochloremia, hypokalemia, metabolic alkalosis, and hypercalciuria. The underlying disorder is incomplete absorption of sodium, chloride, calcium and to some extent potassium. It can cause a mutation in the genes coding for the four proteins involved in sodium chloride reabsorption: Na-K-2Cl cotransporter Type 1 [47], renal potassium channel Type 2 [48], renal chloride channel, ClC-Kb Type 3 [49], and Barttin or mutation in both chloride channels, ClC-Ka and CIC-Kb Type 4 [50]. All are present early in life and types 1, 2, and 4 often in infancy. Type 4 is also associated with deafness. Defective chloride reabsorption impairs paracellular calcium reabsorption and leads to hypercalciuria. A gain of function mutation in the calcium ion-sensing receptor can cause a Bartter like syndrome Type 5 since this receptor inhibits sodium chloride reabsorption when it is activated. It is inherited as an autosomal dominant trait which is also associated with hypocalcemia [51].
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis is a rare autosomal recessive disorder caused by a mutation in either CLDN-16 [52] or CLDN-19 [53] genes which code for claudin 16 and 19. Affected individuals have polyuria, hypermagnesuria, hypercalciuria, tetany, recurrent nephrolithiasis and progressive loss of renal function from the nephrocalcinosis. The urinary loss of magnesium leads to hypomagnesemia, hypocalcemia and tetany. Mutations in claudin 19 also cause ocular abnormalities.
Familial juvenile hyperuricemia is a rare autosomal dominant disorder characterized by hyperuricemia from defective uric acid excretion, gout and progressive renal failure. Medullary cystic disease type 2 is probably the same disease. Most of the patients with these disorders have a mutation in the UMOD gene which codes for uromodulin Tamm-Horsfall protein [54, 55].
5. DISTAL CONVOLUTED TUBULE (FIG. 4, TABLE 6)
Gitelman syndrome is an autosomal recessive disorder characterized by hypomagnesemia, hypokalemia, metabolic alkalosis, and tetany [56, 57]. It is caused by a mutation in the SLC12A3 gene that codes for the sodium-chloride symporter or the thiazide-sensitive Na+-Cl- cotransporter [NCCT] [58]. Affected individuals usually present in late childhood or even not until adulthood with tetany. They also have hypocalciuria in contrast to Bartter syndrome patients.
The hypomagnesemia is due to down-regulation of the epithelial TRPM6 channel in the distal convoluted tubule.
6. CORTICAL COLLECTING DUCT (FIG. 5, TABLE 6)
6.1. Hypertension
Liddle syndrome is an autosomal dominant causing hypertension with variable degrees of hypokalemia and metabolic alkalosis [59]. It is caused by a mutation in either SCNN1B or SCNN1G gene which codes for the β or γ subunit of the epithelial sodium channel ENaC [60]. ENaC mediates sodium reabsorption in the cortical collecting duct. The mutated protein is resistant to degradation and the channel remains open, longer than normal in the luminal leading to excessive sodium reabsorption, volume expansion, and hypertension. The excessive sodium reabsorption increases luminal negativity and increased potassium and hydrogen excretion. The volume expansion causes renin and aldosterone suppression which distinguishes Liddle syndrome from hyperaldoteronism.
Apparent mineralocorticoid excess AME is an autosomal recessive disorder that has signs and symptoms similar to Liddle syndrome [61]. It is cause by a mutation in the HSD11B2 gene that codes for the enzyme 11β-hydroxysteroid dehydrogenase [62,63]. 11β-Hydroxysteroid dehydrogenase mediates the intracellular inactivation of cortisol preventing it from activating the mineralocorticoid receptor and increasing sodium reabsorption and hydrogen and potassium excretion. Like Liddle syndrome, AME is associated with low levels of renin and aldosterone.
Gordon syndrome or pseudohypoaldosteronism type II is another genetic cause of hypertension [64,65]. It is the clinical inverse of Gitelman syndrome with hypertension, hyperkalemia, and metabolic acidosis and is caused by a mutation in either the WNK4 or WNK1 gene which code for the WNK4 protein and WNK1 protein. In the distal convoluted tubule, WNK4 down-regulates NCCT and a mutated WNK4 leads to increased NaCL absorption. In the cortical collecting duct, WNK4 down-regulates the ROMK potassium excretory channel and the mutated protein inhibits more strongly. WNK1 inhibits WNK4 and mutations, the WNK1 gene causes increased expression and enzyme activity.
6.2. Normotension to Hypotension
Autosomal dominant pseudohypoaldosteronism type I PHA1 is a rare disorder caused by a mutation in the gene - MLR - which codes for the intracellular mineralocorticoid receptor [66]. The mutated receptor is unable to react with aldosterone, leading to defective sodium reabsorption and potassium and hydrogen excretion. Salt wasting in autosomal dominant PHA1 tends to be mild and diminishes with age.
Autosomal recessive inheritance PHA 1 is cause by a mutation in either the SCNN1A, SCNN1B or SCNN1G genes which code for the α, β, and γ subunits of ENaC [67]. Infants with this disorder present with severe sodium wasting, hyperkalemia, and metabolic acidosis. They also may have transient tachypnea of the newborn and recurrent respiratory infections.
6.3. Genetic Distal Renal Tubular Acidosis dRTA
Autosomal dominant dRTA is caused by a mutation in the gene - AE1- that codes for the basolateral anion exchanger in the collecting duct intercalated cell [68,69]. Defective anion exchanger function inhibits bicarbonate transport out the cell causing cellular alkalinization and inhibition of apical hydrogen ion secretion. Affected individuals with dRTA have metabolic acidosis, hypokalemia, hypocitraturia, nephrolithiasis and nephrocalcinosis.
Autosomal recessive RTA is cause by a mutation in either the gene ATP6V1B1 [70], encoding the B-subunit of the apical h [+] ATPase or the gene ATP6V0A4 [71], encoding the a-subunit of this protein. Affected individuals with either mutation have the problems associated with dRTA and patients with a mutation in ATP6V1B1 have progressive deafness [72].
6.4. Nephrogenic Diabetes Insipidus [NDI]
Most patients with NDI have a mutation in the gene AVPR2 on the X chromosome [73]. AVPR2 codes for the arginine vasopressin receptor that responds to circulating vasopressin. Activation of this receptor leads to the insertion of aquaporin 2 into the luminal membrane facilitating water transport from lumen of the collecting duct. Autosomal recessive NDI is caused by mutations in the gene AQP2 which codes for aquaporin 2 [74].
7. GENETIC CYSTIC KIDNEY DISEASE (TABLE 7)
7.1. Polycystic Kidney Disease
There are a number of renal diseases, both genetic and congenital, that are associated with multiple cysts one or both kidneys. However, the term, polycystic kidney disease, is reserved for two specific hereditary kidney diseases, autosomal dominant ADPKD and autosomal recessive polycystic kidney disease ARPKD. The most common form of polycystic kidney disease manifesting in children is ARPKD [75,76].
ADPKD typically presents in adults, but can be seen in children and rarely even in neonates [77,78]. It occurs in 1 out of 400-2000 live births, although it is estimated that only half of affected patients will be diagnosed as many patients have clinically silent disease. Approximately 86% of affected families have an abnormality in the PKD1 gene which codes for polycystin1 [79]. Most of the remaining families have a mutation in PKD2 gene which codes for polycystin 2 [80]. While it is typically clinically silent in children, 60% of children with a known mutation have ultrasonically detectable cysts. Affected individuals usually have an increasing size and numbers of cysts over years, leading to massive nephromegaly and renal failure. Hepatic cysts are also common, especially in women.
ARPKD, previously called infantile polycystic kidney disease, typically presents in infancy, although there are childhood and adolescent forms which are generally less severe. It is caused by a mutation in the PHKD1 gene which codes for fibrocystin [81]. The cyst are small to microscopic and due to collecting duct ectasia. The estimated incidence is 1:10,000 to 1:40,000 children. ARPKD is always associated with some degree of hepatic fibrosis. About a third of affected infants will present in the neonatal period, another third of patient with ARPKD will present after infancy, and another third may not present until after the age of 20 years.
7.2. Nephronophthisis
Nephronophthisis is a group of rare autosomal recessive disorders characterized by progressive renal failure associated with tubulo-interstitial fibrosis [82-95] and is considered a “ciliopathies”. It is estimated that nephronophthisis accounts for about 2-10% of end-stage renal failure in children. The symptoms of nephronophthisis are non-specific and include: polyuria/polydipsia and recurrent dehydration. Mutations in at least 13 different genes have been associated with nephronophthisis and listed in table. Depending on the gene mutation, nephronophthisis may be associated with eye, liver and neurological disorders. The proteins from the genes associated with nephronophthisis localize to primary cilia, centrosomes, and/or adherens junctions of renal tubular cells and play an important role in cell-cell and cell-matrix signaling.
CONCLUSION
It is increasingly clear that many renal disorders in pediatrics are a consequence of genetic mutations. In the future, genetic testing will become as easy and as common as ordering a serum creatinine is today.
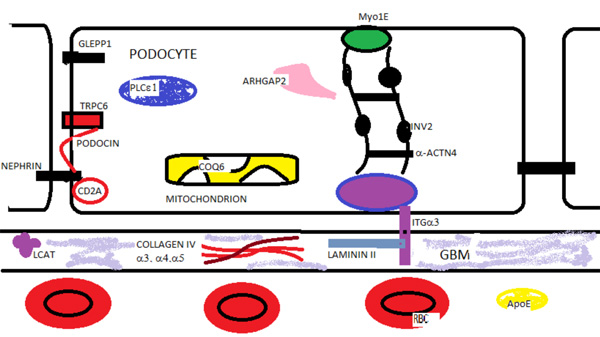
Glomerular Proteins in Hereditary Renal Disease. Nephrin, podosin, PLCε1, Myo1E, Arhgap2, GLEPP1 (PTPRO) are podocyte proteins associated with autosomal recessive nephrotic syndrome. TRPC6, INF2, and α-ACTN4 are proteins mutated in autosomal dominant nephrotic syndrome. Mutations in the mitochondrial enzyme co-enzyme Q6 (COQ6) and the α-3 subunit (ITGα3) of the transmembrane integrin are also associated with nephrotic syndrome. Mutations in the glomerular basement membrane (GBM) proteins - collagen IV α 3,4,5 chains and laminin II cause renal disease. Lecithin-cholesterol acyltransferase deficiency leads to lipid deposits in the GBM and renal disease. Laminated apolipoprotein (APOE) deposits are found in the capillary in lipoprotein glomerulopathy.
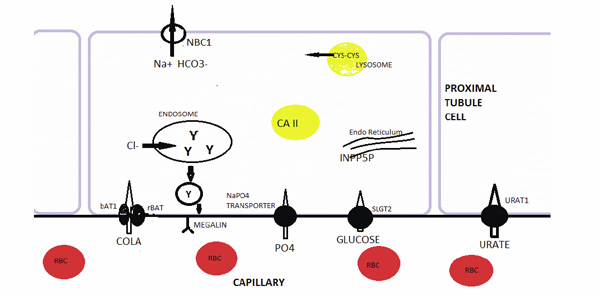
Hereditary Disorders of the Proximal Tubule. Hereditary renal glucosuria is caused a mutated sodium-glucose transporter SLGT2. A mutated bAT1 or rBAT protein, which make up the luminal cystine and dibasic aminoacid transporter leads to cystinuria. Proximal RTA can be caused by a mutated carbonic anhydrase II (CA2) or the basolateral membrane sodium-bicarbonate transporter (NBC1). Hereditary hypophosphatemic rickets is caused by a mutated sodium-phosphate transporter in the luminal membrane. Hereditary hypouricemia is caused by a mutated urate transporter URAT1. A mutated cystinosin in the lysosomal membrane leads to cystine storage and Fanconi syndrome (FS). Mutated phosphotylinositol-4,5-biphosphate 5-phosphatase (INPP5P) in the Golgi apparatus leads to Lowe syndrome and FS. FS is also caused by a mutation in the CLC-5 chloride channel in the endosome leading to impaired recycling of megalin.
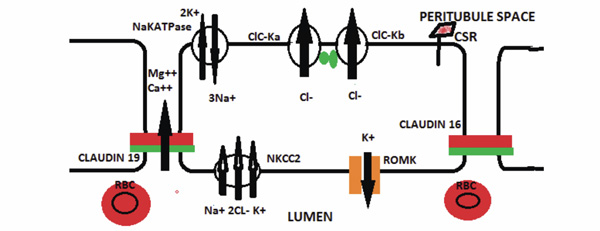
Hereditary Disorders of the Thick Ascending Limb Cell. The major hereditary disorder of the ascending limb of the Loop of Henle is Bartter syndrome which can be caused by a number of mutated proteins. An abnormality in the NKCC2 protein impairs sodium, potassium, and chloride reabsorption from the lumen. A mutated ROMK protein can also cause Bartter syndrome by impairing potassium recycling for the NKCC transporter. Bartter syndrome can also be caused by a mutated CIC-Kb, Barttin ( ), or both CIC-Ka and CIC-Ka. A mutated calcium sensing receptor (CSA) that is constitutively on can cause Bartter syndrome by down-regulating the NKCC2 in association with hypocalcemia. Impaired luminal calcium absorption causes decreased paracellular calcium absorption and hypercalciuria. Familial hypomagnesemia, hypercalciuria with nephrocalcinosis syndrome is caused by either a mutated claudin 16 or 19 protein that impairs paracellular calcium and magnesium absorption.
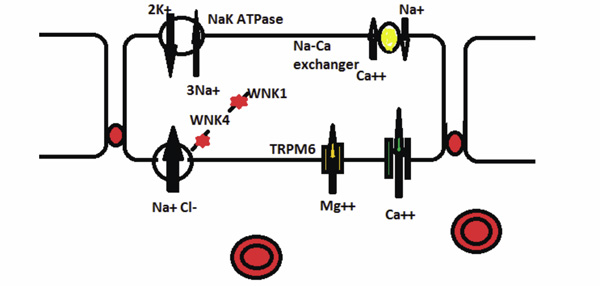
Hereditary Disorders of the Distal Convoluted Tubule (DCT). Gitelman syndrome is caused by a mutated electroneutral sodiumchloride transporter. The decreased NaCl absorption impairs magnesium absorption through the TRPM6 channel. WNK4 inhibits the NaCl transporter and decreases NaCl absorption. A mutated WNK4 in Gordon syndrome leads to increased NaCl absorption in the DCT. WNK1 inhibits WNK4. A mutated WKN1 inhibits more strongly increasing NaCl absorption in Gordon syndrome.
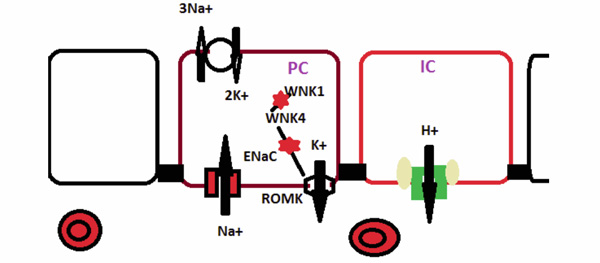
Hereditary Disorders of the Collecting Duct. Sodium enter the principal cell via the epithelial sodium channel (ENaC) which is composed of the α. Β, and γ subunits. In Liddle syndrome mutated β or γ subunit keeps the channel open longer and increases sodium absorption. In pseudohypoaldosteronism, a mutated α, β, or γ subunit impairs the function of the ENaC. WNK4 protein inhibits ( ) potassium secretion via the ROMK transporter. In Gordon syndrome, a mutated WNK4 protein more strongly inhibits ROMK activity. In the intercalated cell (IC) distal RTA is caused by either a mutated anion exchanger (AE1) or either the ATP6V1B1 ( ) or the ATP6V0A4 ( ) subunit of the H+-ATPase.
ABBREVIATIONS
CONFLICT OF INTEREST
The author confirms that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

