
RESEARCH ARTICLE
The Pathophysiology of Antiphospholipid Syndrome
Pablo Ruiz Sada1, Hannah Cohen 2, David Isenberg3, *
Article Information
Identifiers and Pagination:
Year: 2015Volume: 8
Issue: 2
First Page: 2
Last Page: 9
Publisher ID: TOUNJ-8-2
DOI: 10.2174/1874303X01508010002
Article History:
Received Date: 2/7/2014Revision Received Date: 1/9/2014
Acceptance Date: 2/9/2014
Electronic publication date: 20/2/2015
Collection year: 2015

open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
Abstract
Advances in our knowledge of the pathogenic mechanisms of antiphospholipid syndrome have been achieved in the past few years. Apart from the well-known role of anti-β2-glycoprotein I antibodies, complement, endocrine and genetic factors and a variety of other molecules are now under investigation. These new approaches should lead to novel explanations and potential new treatment options.
INTRODUCTION
The antiphospholipid syndrome (APS) is an autoimmune systemic disorder characterized by thrombosis and obstetric morbidity associated with persistent antiphospholipid antibodies (aPL) [1, 2]. Clinically, it causes venous and/or arterial thrombosis affecting blood vessels throughout the body, resulting in significant morbidity and occasionally mortality. The obstetric manifestations include three or more consecutive unexplained miscarriages, one or more unexplained death of a normal fetus at or beyond the 10th week of gestation, evidence of placental insufficiency or one or more premature birth of a normal neonate before the 34th week of gestation because of eclampsia or preeclampsia, or evidence of placental insufficiency [1] Serologically, the standard tests for APS are the lupus anticoagulant (LA), IgG and/or IgManticardiolipin (aCL) or IgG and/or IgManti-β2-glycoprotein Iantibodies (anti-β2-GPI) which to diagnose the patient must be present on two or more consecutive occasions at least 12 weeks apart. The International consensus (revised Sapporo) classification criteria state that APS is present if at least one each of the clinical criteria and one of the laboratory criteria are met (Table 1)
Revised classification criteria for APS
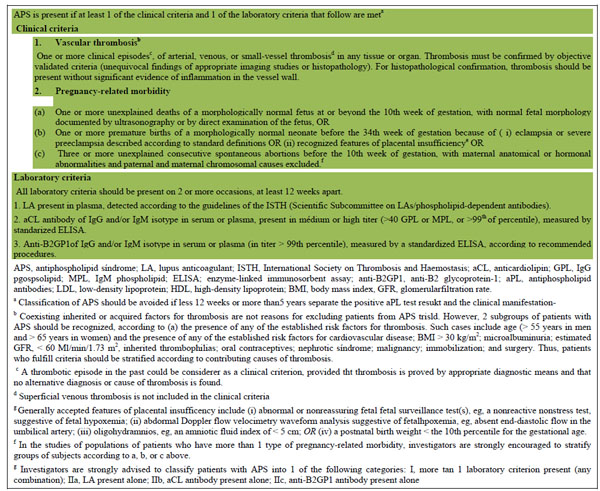 |
Additional clinical manifestations which are not included in the classificarion criteria (referred to as non-criteria manifestations) may be observed such as livedoreticularis, neurological manifestations, nephropathy, thrombocytopenia and heart valve disease even though they are not included in the classification criteria. The kidney is a major target organ in APS. However, it has not received much attention because of the common association between APS and systemic lupus erythematosus (SLE), which has historically, focused studies on immune-complex-mediated glomerulonephritis rather than renal vascular lesions [3]. There are many clinical presentations, due to both large vessels (arterial and venous) and microvasculature involvement (Table 2).
Clinical manifestations of renal involvement in APS.
| Vascular Lesion | Clinical Consequences |
|---|---|
| Renal artery lesions | Arterial renovascularhypertension, renal infarcts |
| Glomerular capillarythrombosis | Tendency to renal insufficiency |
| Renal thromboticmicroangiopathy | Systemic hypertension and renal failure |
| Renal vein thrombosis | Renal failure |
Catastrophic APS (CAPS) is a rare, acute-onset condition that is characterized by sudden extensive microvascular thrombosis leading to multiorgan failure, most frequently affecting the kidneys (71%), as well as the cardiorespiratory and central nervous systems [7]. CAPS may complicate established APS or present de novo. In an observational study of 1000 APS patients from 13 European countries spanning 10 years, CAPS occurred in 0.9% of patients, with a mortality rate of over 50% [2]. In those who survive, chronic renal failure is not uncommon. The pathogenesis of CAPS is not clearly understood, but episodes are often triggered by infections, surgery, or trauma, and widespread complement activation may play a role. Management is mainly based on case reports and usually comprises therapeutic anticoagulation in combination with immunonodulatory therapy including intravenous immunoglobulin, plasma exchange, corticosteroids, cyclophosphamide and rituximab, and ecululizumab (see below).
SEROLOGICAL CONSIDERATIONS
APS is characterized by antibodies directed against protein epitopes combined to phospholipids; they do not bind directly to anionic phospholipids [8]. Antiphospolipid antibodies were first detected in sera from patients with syphilis, as the assay for the detection of this illness (VDRL, RPR) targets an antigen that contains cardiolipin [9]. The recognition that the real targets for the “so-called” phospholipid antibodies were protein co-factors, notably β2-GPI, was a major step forward. We now distinguish between β2-PI-dependent aCL antibodies that are correlated with thrombosis and fetal loss, and β2-GPI-independent aCL antibodies that are not always present in APS, may be found in infections and seem to be more innocent [10]. Reports suggest that in genetically predisposed individuals, some infections can induce the formation of anti-β2-GPI antibodies, and peptide epitopes of infectious agents can act as antigens and induce in mice aPL [8].
Anti-β2-GPI antibodies can have lupus anticoagulant activity [11, 12]. LA are antibodies directed against plasma proteins such as ß2-GPI, prothrombin, or annexin V that are bound to anionic phospholipids [13]. The detection of LA relies uponfunctional clotting tests. There are three reasons to call the term LA a misnomer. LA is associated with a thrombotic tendency rather than an anticoagulant effect and in fact, the anticoagulant is only an in vitro observation. Just about 50 percent of individuals with LA meet the American College of Rheumatology criteria for the classification of SLE, and there is more than one antibody associated with lupus anticoagulant activity. LA activity due to anti-β2-glycoprotein Iantibodies correlates more strongly with thrombotic complications [14, 15]than LA caused by antiprothrombin antibodies. The presence of LA is a stronger risk factor for the development of thrombosis than are aCL oranti-β2-GPI [16].
Antibodies against β2-GPIare found in a large percentage of patients with APS [17]. They target a plasma protein, also known as apolipoprotein H, which has unknown function. Despite many studies suggesting itis a lipopolysaccharide scavenger protein or complementregulator, several researchers suggest that lack of ß2-GPIis not associated with any disease phenotype [18, 19] and can act as a naturally inhibitor of coagulation and platelet aggregation [7, 20]. This haemostasis inhibitor role of ß2-GP-Icould explain why neutralizing antibodies promote thrombosis. It is known that among the population of ß2-GPI antibodies, those specifically directed against an epitope on domain 1 of the protein are the most strongly associated with thrombosis [21].
CONSIDERING POSSIBLE MECHANISMS
The pathophysiology of APS is incompletely understood. Although a prothrombotic basis is presumed, the thrombotic mechanisms are not well established in arterial and venous thrombosis. The obstetric problems are not due to thrombosis alone as thrombosis is neither a universal nor a specific feature in aPL-associated pregnancy loss, [22] and alternative mechanisms are emerging to explain aPL-associated pregnancy morbidity. Even less is known about the non-criteria manifestations, although as suggested by the RITAPS study, B cell depletion is probably effective [6].
A concept called “the two-hit theory” is used to explain the pathophysiology of some diseases. In the case of APS it implies that two triggersare needed to achieve the prothrombotic state. The first hit in the case of APS is likely to be the presence of aPL which have inflammatory and prothrombotic properties in endothelial, dendritic and mastocytic cells. The second would be an acute precipitating event such us surgery, infection, immobilization, pregnancy or oral contraceptives [7]. Autoantibodies from patients with APS potentiate thrombus formation when infused into mice with injured vessels. If the fraction of anti-β2-GPI is removed, the clotting tendency disappears [23, 24].
Although many antibodieshave been involved in the pathogenesis of APS (including those binding to β2-GPI, prothrombin, annexin A5, protein S, protein C, factor X1, and factor XII), [25, 26] β2-glycoprotein seems to be the most relevant target [27]. This protein contains five homologous domains and can present in two different structural configurations. Domain V binds with negative charged phospholipids surfaces, whereas domain I binds antibodies. This first domain of β2-GPI is identified to be the main epitope of the protein. Using domain-deletion mutants of the protein, Iverson et al. found that most anti-β2GPI antibodies reacted with domain I [27] There are several studies suggesting there is a clinical correlation between anti domain I anti-β2-GPI and clinical symptoms, both thrombosis and, interestingly, pregnancy morbidity [28] β2-GPI can be found in plasma in the closed circular configuration. This structure changes after interaction with anionic phospholipids (cardiolipin, apoptotic cells surfaces) to become an open linear configuration that exposes the previously hidden domain I epitope [29] and allows interaction with antibodies (Fig. 1)
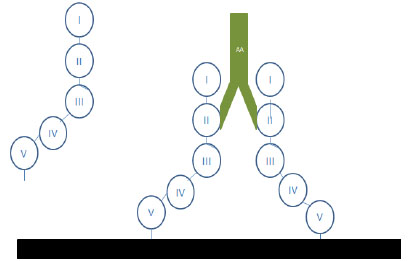 |
Figure 1. β2-GP-I interactions with phospholipids and antibodies |
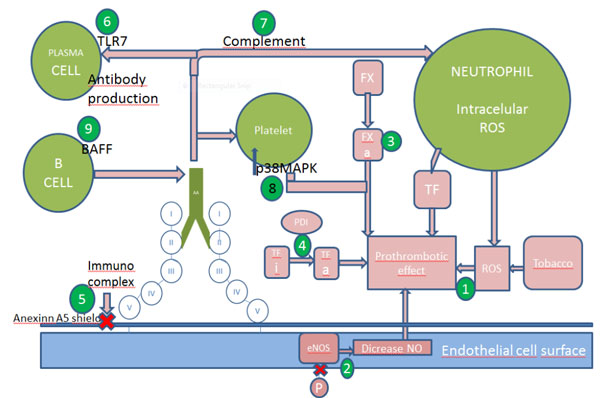 |
Figure 2. Mechanisms of thrombosis and possible targets. Numbers indicates the sites of action of the following drugs: 1 N-acetilcystein, 2 statins, 3 hydroxychloroquine, fluvastatin and FXa inhibitors(anticoagulants e.g. low molecular weight heparin, fondaparinux, rivaroxaban and edoxaban), 4 PDI inhibitors and hydroxychloroquine, 5hydroxychloroquine, 6 TLR7 inhibitors, 7 heparin, eculizumab, 8 belimumab |
. After the formation of the antigen-antibody inmunocomplex, the open linear configuration, also known as the “fishhook” remains open. The complex can activate cellular elements (monocytes, endothelial cells and platelets), inhibit the fibrinolytic system, activate the coagulation cascade, and the complement system [30]. Although the circular form has not been found in plasma, there is in vitro evidence of its presence. Administration of protein H (derived from S pyogenes) to mice has induced anti-β2-GPI [31]. The chemical structure of β2-GPI contains several disulfide bridges which link their domains between each other. In healthy individuals, these bridges are broken, the protein is less immunogenic, and is referred as the “free thiol form”. APS patients also appear to have an increase in oxidative stress. Paroxonase activity, which prevents oxidation of low density lipoprotein cholesterol, is decreased; and [32, 33] 8 epi prostaglandin F, which is a biomarker of lipid peroxidation, is raised [34]. Additionally, monocytes from APS patients have an increased amount of intracellular reactive oxygen species [35]. In these conditions, disulphide bonds are formedand β2-GPI, becomes more immunogenic [36]. Evidence exists that patients with APS have a greater proportion of the oxidized versus the “free thiol form” than do patients with either other autoimmune diseases but without APS, patients with thrombosis without aPL or healthy volunteers (p<0,001 for all comparisons) [36]. In vitro studies showed that the “free thiol form” protects the endothelium from reactive oxygen species (ROS) [35] and that aPL promote formation of intracellular ROS. Furthermore, mice models studies suggest that ROS participate in the genesis of murine thrombosis [37]. Thus, drugs with an antioxidant effect might be helpful in the treatment of APS. There is evidence of N-acetylcysteineinhibition of ROS-mediated thrombosis [38] and coenzyme Q 10 inhibition of aPL mediated ROS generaration [39].
A possible mechanism of thrombosis is aPL-induced damage to the endothelial nitric oxide (eNO) synthesis pathway. APS patients have low plasma levels of eNO compared with controls [40] They also have an impaired, vascular relaxation response due to NO [41]. Endothelial nitrite oxide is synthesized by the conversion of L-arginine by eNOS [42]. The impairment of enzyme activity leads to generation of superoxide and peroxynitrite [43]. In a murine model ß2-GPI antibodies antagonize the activity of endothelial nitric oxide synthase (eNOS) which leads to low NO levels and monocyte adhesion to endothelial cells. Mice deficient in eNOS do not have this antiphospolipid-antibody thrombotic mechanism [44]. In vitro studies also showed HDL cholesterol dependent inhibition of eNOS in women with aPL [45]. Therefore, statins, which up regulate NOS activity, could be a logical treatment option as shown in vitro [46] and in an animal model [47].
Another target under scrutiny is tissue factor (TF). This factor is the key initiator of blood coagulation. After vessel injury, the TF: factor VIIa complex activates the coagulation protease cascade, which leads to fibrin deposition and activation of platelets. Tissue factor is located in an inactive form in the endothelial cell surface, and it needs phosphatidylserine to be exposed after a vessel injury, to be activated and to initiate coagulation. Tissue factor upregulation has been proposed as an important mechanism for development of a prothrombotic state in APS [48], Several studies suggest that patients with APS have increased levels of soluble TF and antibodies against TF pathway inhibitor (TFPI), suggesting impaired downregulation of TF [49, 50]. Further investigation is required to define TF pathway inhibitors. Tissue factor interaction with protein-disulfide isomerase (PDI), an extracellular regulator of thiol exchange, is also necessary to enable the prothrombotic role of this molecule [51]. Its expression is increased in patients with APS, and in vitro studies have demonstrated upregulation induced by antibodies on endothelial cells, neutrophils, monocytes. Potential thaeraputic modalities are PDI inhibitors, which attenuate murine thrombosis, and statins, which inhibit thrombosis in a murine model [52].
As explained above, aPL can upregulate the expression of procoagulant and proadhesive cell-surface molecules such as TF [52]. It is known that this effect is due to the interaction between ß2-GPI antibodies between different receptors. Anti-ß2-GPI target many different molecules on surface of cells. On endothelial cell surfaces, these antibodies bind proteins such as annexin A2, Toll Like Receptor 4 (TLR4), calreticulin and nucleolin to transfer intracellular signals. In monocytes, ß2-GPI is situated in the raft with annexin A2 and TLR4 [53-55]. Antibodies against ß2-GPI stimulate them to produce TF and TNF alpha. The most relevant targets on platelets have been proposed to be the ApoE2 receptor [56] and glycoprotein Ibalpha [56, 57]. Further work is needed to define how antibodies penetrate into the cell [58] Once antibodies from patients with APS enter monocytes and endothelial cells, they can disrupt mitochondrial function, leading to the generation of ROS and the subsequent expression of TF [59]. Studies in animal models have demonstrated that annexin A2 knockout mice [60] and apoE receptor knockout mice [61] are protected from associated thrombosis with aPL. It is known that A1, an analogue of apoE R2 inhibits ß2-GPI antibodies effects in vivo and in vitro [62, 63]. Furthermore, treatment with NAC and coenzyme Q10 prevented the upregulation of TF related to presence of aPL [64]. A significant increase has been reported in the expression of GPIIb/IIIa on platelets treated with aPL antibodies and a thrombin receptor peptide agonist (TRAP). Treatment of platelets with aPL antibodies results in a significant increase in p38 mitogen-activated protein kinase (p38MAPK) phosphorylation, and aPL-induced platelet aggregation and thromboxane B2 (TXB2) production was abrogated by SB203580 (a p38MAPK inhibitor) [65].
Factor X, is an enzyme of the final common pathway of the coagulation cascade which needs to be in its active form, FXa, to exert its procoagulant effect. In APS, FXa reactive IgG has higher avidity binding to FXa, as well as a greater procoagulant effect, than in SLE patients without APS. FXasignalling is linked to interaction with protease-activated receptor (PAR) which leads to the release of intracellular calcium. Recently a study with 36 patients, including controls, characterized the interaction between FXa and human umbilical vein endothelial cells (HUVEC). An improvement was observed in FXa stimulation of HUVEC mediated via enhanced by FXa-reactive APS-IgG and a reduction of intracellular Ca2+ release in cells treated with a specific FXa inhibitor, HCQ and fluvastatin. These results suggest the important role of anti-FXaIgG predicting response to treatment with FXa inhibitors in APS [66].
APL appears to reduce annexinA5 levels and accelerate coagulation of plasma on cultured trophoblasts and endothelial cells. AnnexinA5 forms clusters that bind with high affinity to the surface of anionic phospholipids such as phosphatidylserine, and forms an anticoagulant shield made up of two-dimensional crystals that block phospholipids from availability for coagulation enzyme reactions. Antiphospholipid (aPL) antibodies cause gaps in the ordered crystallization of annexin A5, which expose phospholipids and thereby accelerate blood coagulation reactions. The reduction of annexinA5 levels on vascular cells may therefore be an important mechanisms leading to thrombosis and pregnancy loss in APS [67]. An ex vivostudy demonstratedthat plasmas with anti-ß2-GPI-dependent LAC that recognize domain I displayed significantly increased annexinA5 resistance. These observations suggest aPL may promote thrombosis by interfering with the anticoagulant activity of annexin A5 [68]. Endothelial cells exposed to aPL showed a decrease inannexin A5. The use of hydroxychloroquine (HCQ) is associated with inhibition of the annexin A5 shield in vivo,[69] and a decrease in thrombosis due to APS in mice [70].
Complement activation by aPL has recently been shown in mice models to play an important role in thrombosis, pregnancy loss and fetal growth restriction. APS mouse models demonstrate that passive transference of aPL from APS patients, to pregnant mice induce fetal loss and intrauterine growth restriction [71]. An association between thrombosis and complement in APS has also been demonstrated. Mice with and without C5 and C3 received both aPL and a vessel injury and only thrombi were formed in C5+ and C3+ mice. Complement is a complex system of plasma protein precursors. After a stimulus they become active and resulting in a stepwise cascade [72]. One of these proteins, C5, activated by aPL, leads toactivation of neutrophils and release of TF [73]. Thus, C5 and other complement proteins (C3) have been proposed as possible targets [74, 75]. Reports document the use of eculizumab, a humanized IgG monoclonal antibody that inhibits C5 cleavage and thereby generation of C5a and C3b, to treat catastrophic APS [76] and to prevent to prevent thrombosis after renal transplantation in patients with a history of CAPS [77]. In addition, an ongoing phase 2 study sponsored by Johns Hopkins University should be completed in 2015 (ClinicalTrials.gov Identifier: NCT01029587). Because heparin inhibits complement activation [78], mice treated with heparin after the injection of aPL do not develop fetal loss, unlike mice treated with the direct thrombin inhibitor, hirudin [79]. Some authors suggest that heparin prevents fetal loss through its effect on the complement pathway rather than an anticoagulant effect [80]. Although the current recommendations for APS patients are initial low molecular weight heparin (LMWH), LMWH has not been tested in patients with APS, and data from mice models are unclear [79].
Antiphospholipid syndrome occurs in 10-15% of patients with SLE although, 30-40% have aCL [2]. In addition, approximately 35% of patients with SLE have LA [80] and ß2-GPI [81] antibodies respectively. In a mice model of SLE, the spontaneous development of domain 1 antibodies and analogous APS supported a relationship between the two syndromes and their pathogenesis [82]. Further investigations of this model suggested an important role for the translocation of the TLR7 gene, [83] with leads to increase antibody production [84], aPL can upregulate the expression of TLR7 on plasmocytoid dendritic cells [85]. Inhibition of TLR7 would be a potential target in patients with SLE and aPL.
B cell activating factor (BAFF) is a cytokine necessary for B cell survival which leads to development of an inhibiting antibody to target it [86]. Belimumab, which specifically targets BAFF, has recently been approved for SLE patients with joint and skin disease [87]. In a mice model, belimumab prevents thrombosis [88]. Thus, BAFF may be a potential target in the prevention of thrombosis in high risk patients with SLE.
Female/male ratios of autoimmune diseases invariably show a strong bias to female predisposition, [89] often in the childbearing years, reflecting a probable hormonal influence on their aetiology. Ben-Chetrit hypothesized that apparently healthywomen with “silent” illness might develop active disease because of repeated and prolonged exposure to estradioland gonadotropin [90]. Bruce and Laskin suggested thatacute hyperestrogenemia would be a risk to SLE patient. In a cohort of 1000 APS patients, Cerveraet all found a female/male ratio of 5:1. This ratio was higher in patients with SLE (7:1) than in primary APS (3,5:1) (p< 0,005) [91]. However, in an American retrospective studio with non-SLE associated APS in which patients received ovarian stimulation, there were no reports of thrombosis or maternal complications [92]. Studies to improve knowledge of hormonal influence are in progress.
It is becoming increasingly clear that the concurrence of environmental factors with genetic abnormalities or genetic predisposition, determines whether and when a person will develop APS [93]. Several studies suggest an increased risk of venous thromboembolism(VTE) linked to the presence of heritable thrombophilic defects with aPL. In a Dutch study, Factor V Leiden and the G20210A prothrombin mutation were shown to contribute to the risk of VTE in patients with SLE, with the risk potentiated when one of these thrombophilic defects occurred in combination with LA and/or aCL [94]. Many other studies have implicated genetic factors. In a series of 23 patients with aCL, 29 of their 87 relatives (33%) also had aCL [95]. In Canadian, German, Italian, and Mexican patients an association of aPL with HLA-DR7 has been observed, and in American and Spanish patients, with HLA-DQ [96]. In a large Swedish cohort of patients with SLE, the expression of various aPL was associated with HLA-DRB1 04 and HLA-DRB1 13. These alleles were also found to be more frequent among patients with cerebral stroke and with any vascular event (venous thromboembolism, ischemic heart disease and cerebral stroke), respectively [97]. Certain polymorphisms of the ß2-GPI gene and of the HLA-DMA and HLA-DMB genes may also enhance the risk of development aPL antibodies after been inherited [98], Further investigations are needed to understand the genetic predisposition to develop APS.
CONCLUSION
In conclusion, it is evident that a variety of mechanisms may act independently, or in conjunction, to lead to the clinical features associated with APS. The challenge is to determine, which of these are the key mechanisms in APS associated with venous and/or arterial thrombosis, and pregnancy loss, as well as the key mechanisms in the non-criteria manifestations of APS.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.



