All published articles of this journal are available on ScienceDirect.

TGF-β Made Easy
Abstract
Renal fibrosis is the final common pathway of several nephropathies including chronic allograft failure. Most chronic renal diseases result in tissue fibrosis, and this is independent of their initial cause. Tissue fibrosis is an accumulation of extracellular matrix, and in animal models of renal fibrosis, mRNA levels for profibrogenic cytokines such as transforming growth factor β (TGFβ) and extracellular matrix (ECM) molecular components are up regulated and they precede glomerulosclerosis and interstitial fibrosis. TGFβ 1 plays a crucial role in renal fibrosis. In this review the main features of this important cytokine and existing previous therapeutic attempts to inhibit TGFβ expression are briefly summarised.
TRANSFORMING GROWTH FACTOR β 1
The TGF β 1 Gene
The TGFβ 1 gene is located on human chromosome 19q13.1-q13.3 and on chromosome 7 in the mouse [1]. The TGFβ 1 precursor gene contains 7 exons and very large introns [2]. (Fig. 1). The 5’-flanking sequence of the TGFβ 1 gene contains 5 different regulatory regions, one with enhancer-like activity, two with negative regulatory activity and two with promoter activity [3]. The negative regulatory regions (-1362 to -1132bp and -731 to -453bp) repress the activity of the transcriptional unit [3]. The enhancer-like activity regions (-1132 to-731bp) overcome the activity of the more downstream negative regulatory region [4]. The first of the promoter regions has a positive regulatory activity (-453 to -323bp) [3]. When this region is abolished there is no transcriptional capacity for the upstream TGFβ 1 promoter. Although the second promoter region (-271 to -1bp) is a major site of initiation of transcription, RNA transcription starts at multiple sites [3]. Sequences downstream from the +1 start site are required for expression of human TGFβ 1 gene and one of the major TGFβ 1 mRNAs is independently regulated and transcribed from the second promoter region [4]. After the two promoters, there is a long untranslated first exon [3].
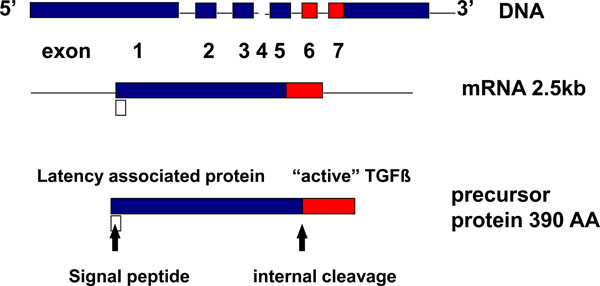
TGFβ: gene to protein.
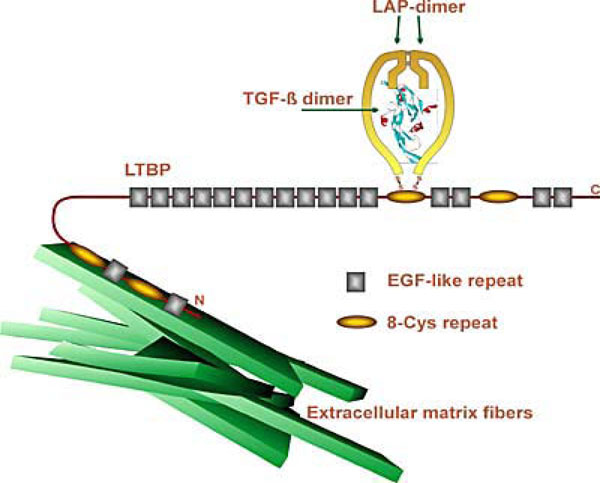
TGFβ latency and its relationship with ECM.
TGF-β1 has the capacity to auto regulate expression of its own mRNA [3]. The TGFβ 1 promoter has two specific regions that are responsive to auto induction [5] one 5’ to the upstream transcriptional start site and another between the two major transcriptional start sites. In both promoter regions, auto induction is mediated by binding of the AP-1 (Jun-Fos) complex. TGFβ 1 auto induction is inhibited if c-jun or c-fos are blocked.
TGFβ 1 mRNA Expression
There are high levels of TGFβ 1 mRNA and/or protein in developing cartilage, endochondral membrane bone, and skin [6]. This identifies the role of TGFβ 1 in growth and tissue differentiation. TGFβ 1 gene transcript is also detected in solid tumour cells and in malignant cells of haematopoietic origin. Normal peripheral blood lymphocytes and placenta also express TGFβ 1 mRNA.
Functional Single Nucleotide Polymorphisms
TGFβ1 is capable of regulating its own gene transcription. Other mechanisms of genetic control include single nucleotide polymorphisms (SNPs) within the 5’ region of the gene. SNPs in this region have been linked to diseases such as arteriosclerosis, bone diseases and several forms of cancer [7]. Grainger et al. demonstrated polymorphisms at position -509 in the promoter are associated with alteration of active and latent TGFβ 1 levels [7]. The SNP at codon 10 is more frequent in blacks compared with whites, and its presence correlated with higher levels of TGFβ 1 mRNA and protein [8]. Mutations in the TGFβ1 gene cause Camurati-Engelmann disease (CED), is a bone sclerosing disorder [9, 10] which is caused by domain-specific mutations of TGFβ1, located in the LAP domain. Mutations in other domains have been found to cause osteoporosis in Japanese women [11].
When TGFβ1 is over expressed this may result in aberrant tissue fibrosis [12, 13]. Fibrosis is the final common pathway of renal disease and solid organ rejection and several studies confirm genetic TGFβ1 polymorphisms [12, 13]. In particular, polymorphisms in the TGFβ 1 promoter were found in graft fibrosis after lung transplantation [14]. There are many other genetic diseases and types of cancer that are originated by genetic mutations in the genes that codify for TGFβ1 receptors and signalling proteins (SMADS) [10].
Distribution of TGFβ 1 mRNA in the Human and Rodent Kidney
TGFβ1 mRNA in Normal Human Kidney
TGFβ1, 2 and 3 mRNAs are weakly expressed in normal kidneys [15]. TGFβ1 protein expression in normal kidneys occurs in the glomerular basement membrane and in the mesangium. TGFβ1 mRNA expression occurs in glomerular cells [16].
TGFβ1 mRNA in Human Glomerular Disease
TGFβ1 mRNA is enhanced in several glomerular diseases [16]. Renal diseases that are not characterised with increased extracellular membrane (ECM) proliferation like thin basement disease and minimal change disease, show the same pattern of TGFβ1 mRNA expression as normal kidneys [17].
Other renal diseases such as: diabetic nephropathy, lupus nephritis, IgA nephropathy, focal segmental glomerulosclerosis and crescentic glomerulonephritis are featured by increased ECM in renal tissue [17]. Here TGFβ1, 2 and 3 mRNAs expression are increased in glomeruli and tubulo interstitium [17].
TGFβ1 mRNA in Chronic Allograft Nephropathy (CAN)
CAN is characterised by tubular atrophy, interstitial fibrosis and a variable degree of glomerulosclerosis. Some authors have measured intragraft expression of TGFβ1 mRNA, and they have found a significant association between TGFβ1 mRNA levels and renal allograft interstitial fibrosis [18].
TGFβ1 mRNA in Rodent Models
The late consequences of diabetic nephropathy are glomerulosclerosis and loss of available filtration surface [19]. There is evidence that high glucose concentration induces TGFβ1 gene expression [19]. Studies in diabetic rats and non obese diabetic mice have shown that TGFβ 1 mRNA levels are elevated in cortical tubular cells [20].
Rats with protein overload have a progressive increase of TGFβ1 mRNA levels in the interstitium and in a lesser degree in cortical tubular cells [21].
In another rat model, fibrosis and interstitial inflammation were produced by a high cholesterol diet and there was significant expression of TGFβ1 mRNA in the renal cortex and interstitium [22].
TGFβ1 mRNA is also expressed in mesangial cells and in resident glomerular cells in rats with Masugi nephritis [23].
TGFβ1 Protein
TGFβ is an extracellular family of proteins that are expressed by most cells [24]. There are three different protein isoforms in mammals (TGFβ1, TGFβ2, and TGFβ3) that have very similar amino acid (AA) sequences and are encoded by three different genes [25]. TGFβ1 is the most widely studied protein in the superfamily, and is the most abundant isoform in cells and tissues.
Protein Structure
The TGFβ1 protein is a homodimer that has a molecular weight of 25 Kδa [25]. Cells secrete TGFβ as a protein complex that is made of three proteins, the mature TGFβ dimer, the TGFβ propeptide dimer or latency associated peptide (LAP), and the latent TGFβ binding protein (LTBP). When mature TGFβ and LAP are separated, TGFβ is activated [25].
Protein Function
TGFβ is a broad-spectrum regulatory cytokine with involvement in embryogenesis, growth, tissue repair and immunological processes [25]. The regulatory properties of TGFβ are in many instances produced by influencing gene expression of other molecules like collagen, fibronectin, tenascin, plasminogen activator inhibitor (PAI-1), and enzymes that inhibit ECM [25].
TGFβ Receptors and Binding Proteins
Most human cells have TGFβ receptors [25] and there are three different types of TGFβ receptors. TGFβ receptors are cell surface proteins. Only receptors II and I are involved in signal transduction. When TGFβ binds to type II receptor, the type I receptor is recruited and phosphorylated to produce a heterodimeric complex that activates signalling pathways [26]. The type III receptor modulates ligand access to the signalling receptors. Receptor I needs receptor II for ligand binding. TGFβ binds first to receptor II and this interaction makes receptor I to be incorporated into the complex and this starts signalling [27]. Type III receptor is a transmembrane proteoglycane and its role is to allow high affinity binding between TGFβ and TGFβ receptor II.
TGFβ Latency
The activity of some growth factors is controlled by their molecules being produced initially in an inactive state requiring downstream activation. Without latency, cytokines would produce their effects before reaching their target cells [28].
The precursor molecule is cleaved in the Golgi apparatus at position 279 after a di-arginine motif by a furin-type protease [28]. The TGFβ propeptide and mature TGFβ are united noncovalently forming a latent complex from which TGFβ must be released to be able to elicit its biological activity. Latency is a critical step in the control of TGFβ activity because TGFβ expression does not always correlate with increased levels of active TGFβ [29]. Latency also regulates TGFβ bioavailability and therefore modulates its function.
Latent TGFβ activation can occur by direct interaction with Thrombospondin-1 (TSP-1). TSP-1 is an adhesive protein that binds to cell surfaces and extracellular matrix.
Mature TGFβ binds to TSP-1 forming a complex in which TGFβ remains active [28].
TGFβ Signalling and Smads Proteins
When TGFβ interacts with cell receptors the signal is transmitted to intracellular signalling cytoplasmic proteins known as Smads. These signalling proteins are transported rapidly into the nucleus and they are able to activate and inhibit functions that mediate the biological effects of TGFβ [30].
TGFβ and Renal Fibrosis
TGFβ has a paramount role in healing and tissue repair. An appropriate balance between extracellular matrix protein synthesis and degradation is essential for growth and healing. Protein degradation is catalysed by several enzymes including plasmin and matrix metalloproteinases (MMP) [31]. When this balance is disturbed fibrosis may result. TGFβ regulates the synthesis of extracellular matrix proteins such as collagen, fibronectin and matrix proteoglycans (Fig. 2). TGFβ is also able to inhibit extracellular matrix degradation by inhibiting plasmin and MMPs [31]. Experiments in animal lung models demonstrate that TGFβ is a potent fibrogenic cytokine that initiates a local fibrotic response that is subsequently perpetuated despite the absence of continued TGFβ expression [31]. In normal kidney tissue, TGFβ mRNA expression is low. However, in proliferative glomerular diseases like mesangial proliferative glomerulonephritis and focal segmental glomerulosclerosis there is excessive regulation of TGFβ. Other non-proliferative glomerular diseases have no increased expression of TGFβ [31]. Several animal models have demonstrated an association between glomerular expression of TGFβ and fibrosis [31]. Border and colleagues used neutralising anti-TGFβ antibody in a rat model of proliferative glomerulonephritis and they were able to show improvement in the glomerular histology [32].
In diabetic nephropathy there is loss of glomerular filtration surface due to glomerulosclerosis and mesangial expansion. Some animal experiments have shown that hyperglycaemia modulates TGFβ gene expression and this effect may be produced in association with other cytokines like IL-1 and platelet derived growth factor (PDGF) [31]. Furthermore, diabetic patients have higher circulating levels and urinary levels of TGFβ than the normal population [31].
Chronic glomerular disease eventually induces interstitial fibrosis and, conversely, chronic interstitial disease may lead to glomerulosclerosis. In renal fibrosis there is an interstitial chronic inflammatory cell infiltrate with proliferation of interstitial myofibroblasts that is cytokine driven [33]. Furthermore, in situations of renal injury, epithelial cells in the kidney may transform into fibroblasts by the process known as epithelial-mesenchymal transdifferentiation [34]. TGFβ and other growth factors and adhesion molecules are involved in this process. The release of TGFβ into the renal interstitium may be produced by renal parenchyma and /or infiltrating monocytes or lymphocytes [31].
The Angiotensin II (AII) interaction with TGFβ has important consequences for renal fibrosis. AII has haemodynamic properties but is also able to act as a growth factor stimulating renal and cardiac cell hypertrophy and increasing expression of type IV collagen mRNA and TGFβ synthesis by cells. Furthermore, administration of anti-TGFβ antibody is able to block the effect of AII on matrix protein synthesis [31]. Experimental models of renal fibrosis based on neutralizing the effects of AII have shown decreased expression of TGFβ [35-37].
TGFβ in CAN
The role of TGFβ in many fibrotic diseases suggested that TGFβ might have significant influence in the onset and progression of CAN. Immunological and non-immunological processes that stimulate aberrant tissue repair lead to fibrosis of the renal allograft. Many factors are involved in the pathogenesis of CAN and their analysis invariably leads to finding up-regulation of TGFβ after renal transplantation [35-37]. Analysis of protocol renal transplant biopsies showed that TGFβ1 expression was linked with the chronic vascular changes seen in CAN [38].
Calcineurin inhibitors have profibrotic effects in the renal allograft and this induction is mediated by increasing TGFβ expression [39]. Renal transplant patients on long-term calcineurin inhibitor treatment express high levels of intragraft TGFβ and this correlates with a decline in renal function [39]. Mohammed et al., analysed renal biopsy specimens from renal transplant patients with decline renal function [40] that were receiving CyA or tacrolimus. These authors found no difference in latent TGFβ expression in the two different treatment groups. However, biopsies from patients receiving CyA showed significantly higher expression of active TGFβ than biopsies of patients receiving tacrolimus. Such difference in active TGFβ expression may reflect a more intense ongoing chronic rejection process in the CyA group but the biopsy findings in these groups of patients with renal transplant dysfunction may be a reflection of events rather than real differences in TGFβ expression induced by the drugs.
The two main limitations of clinical studies evaluating the role of TGFβ in CAN are that they include a reduced number of patients and that some of them don’t distinguish between latent and active TGFβ [35].
Many other cytokines and growth factors have been shown to play a role in CAN. Endothelins are stimulators of extracellular matrix proteins and TGFβ promotes their release from endothelial and tubular epithelial cells. Using the Fischer to Lewis model of chronic rejection, Braun et al. antagonised the endothelin fibrogenic effect and this proved effective in improving histological appearance of rejecting allografts [41].
TGFβ increases rat mesangial cell matrix and stimulates mesangial cell growth in long-term culture [42]. There is some experimental evidence that PDGF up regulation is also involved in this process and this is TGFβ-mediated.
TGFβ INHIBITORS
Inhibition of TGFβ may lead to arrest or reverse renal fibrosis of whatever cause. An important group of inhibitors are proteins that bind TGFβ and prevent its interaction with type I and II receptors. Another ways of inhibiting TGFβ is to use peptides that block its activation, the use of antisense nucleic acids that block TGFβ production, or agents that interfere with the signalling process. TGFβ overexpression underlies human and animal fibrotic diseases and the complexity of this cytokine signalling provides many targets for its blockade with the downside of incomplete TGFβ neutralisation [43].

