All published articles of this journal are available on ScienceDirect.

Methemoglobinemia-Induced Acute Kidney Injury
Authors Info & Affiliations
Abstract
Introduction
Methemoglobinemia refers to the presence of increased levels of methemoglobin (Fe3+) in the blood. Methemoglobinemia can cause cyanosis, dyspnea, fatigue, seizure, arrhythmia, coma, and even death. Although methemoglobinemia is shown to cause acute kidney injury in experimental settings, human case reports are exceedingly rare. In addition, morphological features of methemoglobinemia-induced renal disease in humans remain undefined.
Case Presentation
A 76-year-old man with a history of chronic obstructive pulmonary disease underwent bronchoscopy following local anesthesia with a benzocaine spray. The patient developed benzocaine-induced methemoglobinemia and acute renal failure. Urinalysis disclosed numerous dysmorphic erythrocytes, erythrocyte casts, and granular casts. Urine protein excretion was approximately 1.1 g/day. Serologic tests were negative. Renal biopsy demonstrated minor glomerular abnormalities, severe acute tubular necrosis, and numerous erythrocyte casts in the tubules. Despite supportive care, renal function deteriorated necessitating hemodialysis. Four months later, the patient remained on hemodialysis. To exclude a superimposed pathology, renal biopsy was repeated and showed numerous erythrocyte casts in the tubules and severe tubular damage.
Conclusion
Methemoglobinemia can cause acute kidney injury in humans. Morphological features resemble those observed in methemoglobin-induced acute kidney injury in experimental settings. This case calls for a heightened awareness of potential adverse effects of methemoglobinemia on renal function.
INTRODUCTION
Acute kidney injury (AKI) remains a major cause of morbidity and mortality. AKI due to myoglobin and hemoglobin is well documented. Although methemoglobinemia is shown to cause AKI in experimental settings, human case reports are exceedingly rare. In addition, morphological features of methemoglobinemia-induced renal disease in humans remain undefined. Here, we present a case of methemoglobin-induced AKI requiring long-term renal replacement therapy. Clinical and morphological features of methemoglobin-induced renal disease are presented. Furthermore, the pathogenesis and available treatment options are discussed.
CASE REPORT
A 76-year-old man developed hematuria and acute renal failure following administration of topical benzocaine.
With a past medical history notable for chronic obstructive pulmonary disease, the patient was diagnosed with squamous cell carcinoma of the lung. He had a 40-pack-year history of smoking cigarettes. Medications included an albuterol and ipratropium inhaler as needed. The patient underwent right lower lobe and middle lobe resection. No immediate postoperative complications were noted. Three days after surgery, however, the patient developed hypoxia. Chest radiograph showed atelectasis of the left lower lobe. Following local anesthesia of the pharynx with a benzocaine spray, bronchoscopy was performed and a mucus plug was removed. After the procedure, the patient developed cyanosis. Pulse oximetry showed an oxygen saturation of 67%. Arterial blood gas analysisdemonstrated pH of 7.42, pCO2 40 mmHg, pO2417 mmHg, O2 sat 94%, and bicarbonate 25 meq/liter while thepatient was breathing 100% supplemental oxygen using a face mask. The patient was diagnosed with methemoglobinemia induced by benzocaine. The patient remained hemodynamically stable. Intravenous administration of methylene blue (1 mg/kg) and ascorbic acid (1 mg) resulted in rapid resolution of cyanosis. Over the ensuing days, however, serum creatinine concentration rose to 3.0 mg/dl (Fig. 1). Intravenous fluids were administered for worsening renal function and the renal service was consulted.
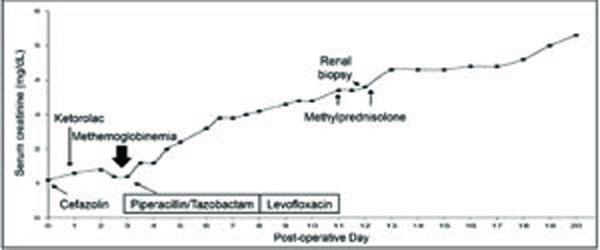
Renal function, diagnostic procedures, and therapeutic interventions during hospitalization.
A review of medical records showed administration of the following medications in the perioperative period: ketorolac (one dose), morphine as needed, pantoprazole, cefazolin (two doses), and piperacillin/tazobactam (Fig. 1). On physical examination, the patient was an elderly Caucasian man in no acute distress. The temperature was 37.2°C, the blood pressure 140/60 mm Hg, the pulse 80 beats per minute, the respiratory rate 12 breaths per minute, and the oxygen saturation 100% while the patient was breathing ambient air. The urine output was 50 cc per hour. The jugular venous distention was within normal limits. The cardiovascular examination was unremarkable. The lungs were clear to auscultation on the left, with diminished breath sounds at the right base. There was a 2+ pitting edema over shins and acne on his back. Laboratory tests showed serum creatinine and urea nitrogen concentrations of 3.0 mg/dl and 37 mg/dl, respectively. White blood cell count was 11,600/μl, hemoglobin 8.2 g/dl, and platelet count 371,000/μl. Urinalysis showed a specific gravity of 1.020, pH 5.0, albumin100 mg/dl, and 3+ blood. Microscopic examination of urine sediment revealed numerous dysmorphic erythrocytes, erythrocyte casts, and granular casts. A protein to creatinine ratio in a random sample of urine indicated proteinuria of approximately 1.1 grams per day.
The clinical presentation and laboratory test results indicated glomerular hematuria and acute tubular necrosis. Despite supportive care including intravenous bicarbonate administration, renal function continued to deteriorate. The patient was pulsed with intravenous methylprednisolone 500 mg per day for suspected rapidly progressive glomerulonephritis. Serological tests for anti-nuclear antibodies, viral hepatitis (hepatitis B surface antigen, hepatitis B surface antibody, hepatitis B core antibody, hepatitis C antibody), anti-glomerular basement membrane antibobodies, and anti-neutrophil cytoplasmic antibodies were all negative. Serum C3 and C4 concentrations were normal. Renal ultrasonography demostrated both kidneys meassuring at 11.8 cm in length. There was no hydronephrosis, cysts, stones, or masses.
Renal biopsy was performed. The sample consisted of renal cortex and medulla. There were 18 glomeruli in the sample submitted for light microscopy. Two glomeruli were globally sclerosed. Another two glomeruli showed chronic ischemic changes. All other glomeruli revealed a normal architecture with normal or slightly increased mesangial cellularity. The glomerular capillaries were patent and glomerular basement membranes were of normal thickness. A rare glomerulus revealed focal capillary congestion (Fig. 2A). Occasional glomeruli revealed erythrocytes in the Bowman’s space and focal epithelial cell hyperplasia (Fig. 2B). Numeroustubules were distended with erythrocyte casts and pigment casts (Fig. 2C, D). Many tubules showed dilated lumina containing necrotic cells. Tubular epithelial cells demonstrated degenerative features such as flattening, vacuolization, and karyorrhexis (Fig. 2C, D). Occasional-tubular epithelial cells contained erythrocytes in the cytoplasm (erythrophagocytosis) (Fig. 2D). The interlobular arteries showed marked intimal fibrosis. Immunofluore-scence microscopy did not reveal IgG, IgA, IgM, C3, C4, C1q, kappa or lambda light chain deposition in the glomeruli. Ultrastructural examination demonstrated degene-rative and regenerative changes of tubular epithelial cells. The glomerular basement membranes were of normal thickness. A histological diagnosis of acute tubular necrosis accompanied by numerous erythrocyte casts was rendered.
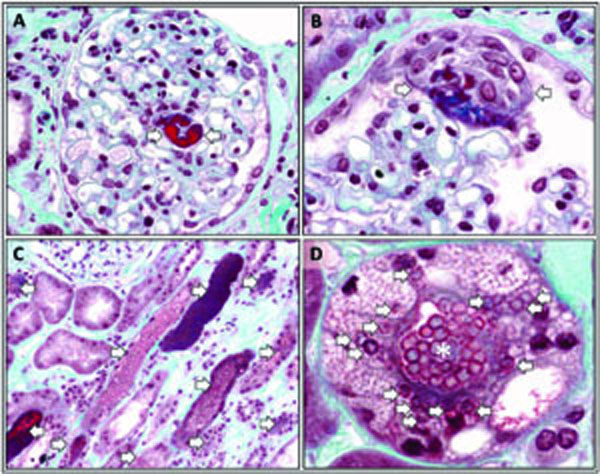
Morphological features of methemoglobin-induced acute kidney injury. A glomerulus with focal capillary congestion or thrombosis (arrows) (A). A glomerulus with focal epithelial cell hyperplasia in the Bowman’s space (arrows) (B). Several tubules packed with erythrocytes (arrows) (C). A tubule packed with erythrocytes in the lumen (asterisk) demonstrating marked vacuolization of the epithelial cells (arrows) (D). Several erythrocytes are seen in the cytoplasm of the tubular epithelial cells (arrows) (D). Tissue sections were stained with trichrome stain (A-D).
Methylprednisolone was discontinued. Despite supportive care, renal function continued to deteriorate necessitating hemodialysis. Four months following discharge from the hospital, the patient still required hemodialyis due to poor renal function. Considering a lack of renal recovery, renal biopsy was repeated. It revealed numerous erythrocyte casts, occasional brown pigmented casts, and severe tubular damage. In addition, there was marked hemosiderin accumulation in the tubular epithelial cells. The interstitium showed patchy fibrosis and scattered mononuclear inflammatory cells.
DISCUSSION
Here, we report a case of benzocaine-induced methemoglobinemia followed by glomerular hematuria and acute renal failure. Renal biopsy demonstrated acute tubular necrosis accompanied by numerous erythrocyte casts in the tubules. On clinical grounds, there was no obvious cause for acute tubular necrosis. In addition, despite the use of penicillins, cephalosporins, and non-steroidal anti-inflammatory drugs (NSAIDs) in the peri-operative period, the renal biopsy did not show NSAIDs- or antibiotic-induced acute interstitial nephritis.
Mammalian hemoglobins consist of four polypeptide subunits (globins) each containing an iron protoporphyrin IX (heme) molecule. When the ferrous (Fe2+) irons contained in the heme molecules are oxidized to the ferric (Fe3+) statemethemoglobin is formed [1]. Methemoglobin does not have oxygen-carrying capacity because the ferric heme is unable to bind oxygen. Under physiological conditions, low levels of methemoglobin are present in the blood accounting for less than 1% of total hemoglobin. Methemoglobinemia refers to the presence of increased levels of methemoglobin in the blood. The formation of methemoglobin may increase when hemoglobin is exposed to endogenous oxidizing agents and free radicals. However, methemoglobinemia of clinical relevance occurs mainly as a result of exposure to certain drugs and toxins. Diamino-diphenyl sulfone (dapsone) and benzocaine are among the most frequently reported causes of acquired methemoglobinemia. The clinical manifestations of methemoglobinemia depend on the methemoglobin level and range from cyanosis, dyspnea and fatigue in mild cases to coma, seizure, arrhythmia, and even death in most severe cases. Individuals with methemoglobinemia may have normal partial pressure of oxygen and normal to falsely elevated calculated oxygen saturation on arterial blood gas analysis. However, pulse oximetry shows reduced oxygen saturation. Thus, a discrepancy in oxygen saturation between arterial blood gas analysis and pulse oximetry may indicate methemoglobinemia. In the present case, the diagnosis of methemoglobinemia was rendered based on the development of cyanosis following benzocaine administration, a discrepancy in oxygen saturation between arterial blood gas analysis and pulse oximetry, and the rapid resolution of cyanosis following methylene blue and vitamin C administration.
Drug-induced methemoglobinemia leading to AKI has been encountered sporadically in humans. For example, an 18-year-old woman who took 2.4 g phenazopyridine over a 24-hour period developed methemoglobinemia, hemolytic anemia, and acute renal failure [2]. Serum creatinine concentration peaked at 9.7 mg/dl and hematocrit decreased to 26%. Because renal failure resolved with supportive care, renal biopsy wasnot performed. Methemoglobinemia-induced acute renal failure was also reported following an overdose of zopiclone, a sedative-hypnotic drug [3]. Renal insufficiency resolved with methylene blue administration and supportive care. Because of the lack of renal biopsy the exact nature of renal injury could not be determined. Finally, acute renal failure was also associated with methemoglobi-nemia due to intoxication with indoxacarb, an insecticide that blocks neuronal sodium channels [4,5]. The nature of renal injury could not be specified. Due to the availability of renal biopsy at the time of diagnosis and again during the follow-up, our case adds to the existing literature on the subject and provides insight into the pathogenesis of methemoglobin-induced AKI in humans.
The pathogenesis of methemoglobin-induced renal injury has been studied in animal models. Intravenous administration of methemoglobin resulted in AKI in experimental settings [6,7]. Kidneys harvested at different time points following methemoglobin administration revealed the following morphological changes: pigment casts in the distal tubules and collecting ducts, tubular dilatation proximal to pigment casts, movement of casts distally, loss of cast pigment, epithelial cell death, and formation of granular casts [7]. The early decline in renal function appeared to be the result of increased viscosity of tubular content and intratubular obstruction by methemoglobin [6]. This was followed by epithelial cell injury and death in the proximal convoluted tubules. Following disappearance of intratubular pigment, non-functioning nephrons with collapsed tubules lined by hemosiderin-laden cells prevailed. In the present case, morphological changes observed in renal biopsy specimens resembled those reported in animal models of methemoglobin-induced AKI including pigment casts in tubules, tubular dilatation, acute tubular necrosis, and extensive hemosiderin accumulation in tubular epithelial cells.
Intratubular pH plays a significant role in tubular toxicity of methemoglobin and hemoglobin [1]. Under physiological conditions, the pH of the ultrafiltrate decreases along the ascending limb of the loop of Henle and reaches its lowest values in the collecting duct. Because acidosis facilitates hemoglobin oxidation, alow pH in the distal nephron can promote methemoglob information and cytotoxicity. This concept is illustrated in the following experiments. To acidify or alkalinize urine, rats were givenan aqueous solution of either ammonium chloride (150 meq/liter) or potassium bicarbonate (150 meq/liter), respectively, instead of drinking water overnight [8]. Aciduric rats infused with methemoglobin or hemoglobin developed acute renal failure accompanied by heme pigment casts in the distal tubules and proximal tubular necrosis. In aciduric rats, methemoglobin resulted in more severe renal injury than hemoglobin. No renal damage was observed in alkalinuric rats infused with hemoglobin. Aciduria promoted methemoglobin formation and precipitation in distal tubules. Obstruction of distal nephron resulted in increased hemoglobin uptake into the proximal tubular epithelial cellsand proximal tubular necrosis. In the presentcase, sodium bicarbonate solution (150 meq/liter) was administered intravenously. However, renal function continued to decline. We argue that this might have been due to the fact that urine was alkalinized several days later, when the injury already set in.
Mannitol is an osmotic diuretic that can scavenge free radicals and cause renal vasodilatation by stimulating renal prostaglandin production. Intravenous administration of mannitol attenuated methemoglobin-induced acute tubular necrosis in rats [7,9]. Zager et al. argued that the protective effect of mannitol on renal function was due to its osmotic diuretic effect [10]. Similarly, the salutary effect of mannitol on renal function in a model of myoglobin-induced AKI was attributed to its osmotic diuretic effect [9]. In the present case, however, intravenous mannitol was not administered.
Hematuria glomerular in origin was another intriguing characteristic of the present case. Although methemoglobin-induced AKI is well documented in experimental settings, acute glomerular injury resulting in hematuria has not been reported previously. With no preexisting renal disease, the patient presented here developed glomerular hematuria confirmed by the presence of erythrocytes in the Bowman’s space and tubules. However, histological examination of renal biopsy specimens demonstrated only subtle glomerular abnormalities that could not account for the large amount of blood present in the tubules. Since the first kidney biopsy was performed nine days after the development of methemoglobinemia, we argue that glomerular injury had resolved by the time tissue sampling was performed.
Erythrophagocytosis was another histological feature in the present case. Tubular epithelial cells are capable of phagocytizing and breaking down erythrocytes and fragments thereof [11,12]. This process is similar to erythrophagocytosis that normally occurs in the mononuclear phagocyte system to remove senescent erythrocytes. To explore whether phagocytosis and digestion of erythrocytes can cause tubular epithelial cell injury, the following experiments were conducted. In an in vivo experiment, autologous heparinized blood was infused into the proximal tubules in the rat. Within six hours, hemoglobin was localized to apical vacuoles and lysosomes of the proximal tubular epithelial cells [11]. Hemoglobin presumably originated from blood hemolyzed during the procedure. Between 6 and 15 hours, there was progressive loss of the brush border and formation of finger-like cell protrusions extending from the surface and surrounding individual erythrocytes in the lumen. At eighteen hours, erythrocytes were localized to the cytoplasm of the proximal tubular epithelial cells. This was followed by marked degenerative changes along with lipid accumulation in the proximal tubular cells. Although this in vivo experiment clearly documented the detrimental effects of hemoglobin and erythrocytes on tubular epithelial cells, erythrophagocytosis did not decrease the viability of cultured human proximal tubular cells [13]. Similarly, acute tubular necrosis was not a universal morphological feature in patients with glomerular hematuria and intratubular erythrocyte casts [12].
The preponderance of evidence suggests that unbound heme and free iron are involved in the pathogenesis of tubular injury observed in glomerular hematuria [14,15]. Heme is an integral component of heme proteins such as hemoglobin, myoglobin, mitochondrial cytochromes, and enzymes that regulate vascular tone, redox reactions, cell signaling, DNA transcription, and RNA translation [16]. In the kidney, hemoglobin is readily taken up by the proximal tubular epithelial cells via megalin and cubilin localized to the brush border [18,19]. Hemoglobin is then dissociated to heme and globin [18]. While globin undergoes proteolytic degradation, heme oxygenases brake downheme into iron, carbon monoxide, and biliverdin [14]. Cellular accumulation of unbound heme can cause protein/DNA denaturation, impaired enzymatic activity, and lipid peroxidation [16]. In addition, heme can induce inflammation by increasing vascular permeability, upregulating the expression of adhesion molecules, and recruiting leukocytes [17]. In vitro studies showed that heme resulted in cell-cycle arrest and apoptosis of cultured proximal tubular epithelial cells in a dose-dependent manner [13, 20]. In addition, hemoglobin and free iron also led to reduced viability of cultured human proximal tubular epithelial cells [13]. In the present case, marked hemosiderin accumulation in the tubular epithelial cells raises the intriguing question whether heme might be involved in the pathogenesis of methemoglobin-induced AKI in humans.
Because of its redox properties, iron can cause oxidative damage by stimulating production of reactive oxygen species [21]. Therefore, the vast majority of iron in the body is bound to proteins preventing it from participating in redox reactions. Nonetheless, a minute amount of iron is found in association with chelators of low molecular mass constituting the labile iron pool [22]. The labile iron can participate in redox reactions and may be involved in the pathogenesis of heme protein-induced acute kidney injury. Of note, rhabdomyolysis resulted in acute tubular necrosis and increased labile iron levels in the kidney of rats [23]. A concomitant reduction in cytochrome P450 enzymes in the kidney led the investigators to conclude that degradation of these heme-containing mono-oxygenases might have contributed to increased labile iron levels in the kidney. Interestingly, administration of deferoxamine, an iron chelator, prevented acute kidney injury induced by myoglobin and hemoglobin in rodents [24,25].
Is methemoglobinemia is a rare event and should one considerits ramifications? Over a five-year period, methemoglobinemia accounted for two thirds of adverse effect reports related to a benzocaine product received by the United States Food and Drug Administration [26]. While over 80% of these adverse events were considered serious, a benzocaine spray was implicated in approximately 90% of the cases. In a retrospective study involving 138 cases of acquired methemoglobinemia, benzocaine accounted for five of the most severe cases with a methemoglobin level of 44% on average [27]. Therefore, methemoglobinemia should be an important consideration when benzocaine products are administered.
CONCLUSION
Methemoglobinemia is a common clinical condition with significant morbidity and even mortality. As documented by this report, methemoglobinemia can result in severe AKI necessitating long-term dialysis. The current case demonstrates that morphological features resemble those observed in methemoglobin-induced AKI in animal models. The pathogenetic mechanisms underlying methemoglobin-induced renal toxicity have been investigated in animal models and generally resemble those responsible for AKI mediated by other heme proteins. In experimental settings, alkalinization of the urine and intravenous administration of mannitol attenuated methemoglobin-induced AKI. However, such measures may be helpful if applied early during the course of disease. A heightened awareness of adverse effects of methemoglobinemia on renal function will facilitate early diagnosis and appropriate treatment.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
G. Warren, MD, assisted in the care of this patient and edited the manuscript.

