All published articles of this journal are available on ScienceDirect.

A Simplified Approach to the Diagnosis of Atypical HUS: Clinical Considerations and Practical Implications
Abstract
Although rare, atypical hemolytic-uremic syndrome (aHUS) carries a high morbidity and mortality. Widespread microvascular thrombosis, thrombocytopenia and microangiopathic hemolytic anemia are the hallmark of aHUS. Virtually any organ (particularly the kidney) can be a target for the devastating effects of this syndrome. Uncontrolled activation of the alternative pathway of the complement system lies at the heart of the pathogenesis of aHUS. While significant advances have been made in our understanding of aHUS, establishing timely diagnosis of this syndrome has been challenging. This, in part, is due to the absence of a sensitive and specific diagnostic test and a relatively lack of our familiarity with the syndrome. With the recent success and approval of a humanized monoclonal antibody (eculizumab) in the treatment of aHUS, prompt and accurate diagnosis is of paramount importance to limit the target organ injury. This article presents a simplified approach to establishing the diagnosis of aHUS.
INTRODUCTION
Atypical hemolytic-uremic syndrome (aHUS) is a thrombotic microangiopathy (TMA). It is characterized by endothelial injury leading to vascular thrombosis [1-3]. Other thrombotic microangiopathies such as thrombotic thrombocytopenic purpura (TTP) and HUS associated with Shiga-toxin producing Escherichia coli (STEC-HUS) also cause vascular thrombosis. While all three diagnostic possibilities must be considered in a patient presenting with TMA, widespread familiarity with TTP (and its successful treatment with plasma therapy) and a conventional wisdom that aHUS is a disease of children lowers the index of suspicion of aHUS on the diagnostic list [3]. In this context, the diagnosis of aHUS can present a diagnostic challenge.
It is worth mentioning that a deficiency of the von Willebrand factor cleaving protease ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) results in TTP. On the other hand, HUS has two different identifiable sources of origin [1-4]. The STEC-HUS is most often seen in the setting of an infection with Escherichia coli (usually E. coli serotypes 0157:H7 and 0104:H4). In contrast, uncontrolled activation of the alternative pathway of the complement system is involved in the pathogenesis of atypical HUS (aHUS) [3]. Of note, there is an intricate reciprocal interplay between complements and the coagulation system and complement activation facilitates vascular thrombosis [5]. In a retrospective autopsy-based study, Hosler et al. conducted a histopathological examination of 56 individuals who succumbed to HUS (n=31) or TTP (n=25) [6]. In individuals with TTP, platelet-rich thrombi were present in the heart, pancreas, kidney, adrenal gland and brain, in descending order of severity. In those with HUS, however, thrombi consisted predominantly of fibrin and red cells and were most abundant in the kidney followed by the pancreas (19%), adrenal glands (13%), brain (6%) and heart (3%). No distinction between “typical” and “atypical” HUS was made in this study, however.
From a simplistic standpoint, there are distinct laboratory tests that are available to make the diagnosis of TTP (ADAMTS13 activity assay) and STEC-HUS (PCR or culture-based examination of a stool sample). However, a single confirmatory diagnostic test for aHUS does not exist. Indeed, microangiopathic hemolytic anemia, thrombocytopenia and target organ injury, particularly kidney damage, are essential clinical features of aHUS.
Microvascular thrombosis in atypical hemolytic-uremic syndrome results in ischemic injury to target organs [5-3]. The kidney is the most common organ involved [3, 13]. In this context, hematuria, proteinuria, hypertension, azotemia and volume overload can all be seen [3, 13]. Important diagnostic elements include thrombocytopenia (platelet count<150,000/ mcL or 25% decrease from baseline), microangiopathic hemolytic anemia (schistocytes on blood film, elevated lactate dehydrogenase, decreased haptoglobin, decreased hemo-globin) and target organ injury (elevated blood urea nitrogen and creatinine, abnormal liver function tests, elevated pancreatic enzyme levels, stroke, myocardial infarction etc.) [3] (Fig. 1).
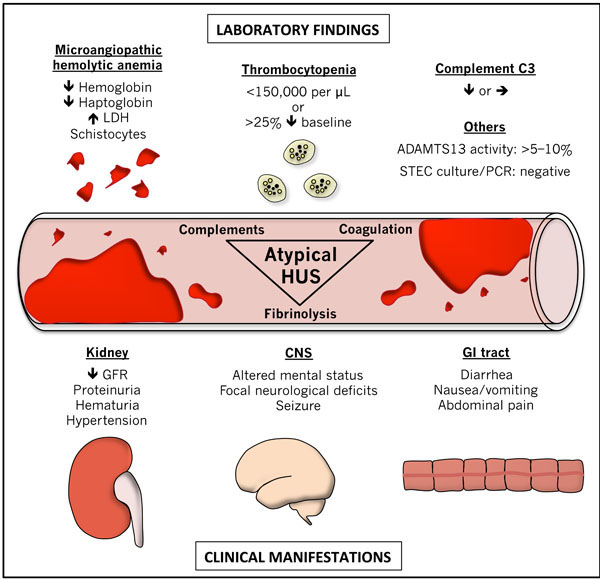
Clinical and laboratory manifestations of atypical hemolytic-uremic syndrome. CNS, central nervous system; GFR, glomerular filtration rate; GI, gastrointestinal; LDH, lactate dehydrogenase; PCR, polymerase chain reaction; STEC, Shiga toxin-producing Escherichia coli; ↑, increased; ↓, decreased; →, unchanged; >, greater than; <, less than.
Laboratory investigations such as Shiga-toxin test and ADAMTS13 activity can be very helpful in distinguishing HUS, TTP and aHUS from one another. A deficiency of ADAMTS13 (less than 5% of normal activity) points to the diagnosis of TTP while the presence of Shiga toxin indicates STEC-HUS. Normal ADAMTS13 activity and absence of Shiga toxin help establish the diagnosis of aHUS in patients presenting with thrombotic microangiopathy. A major limitation of the ADAMTS13 activity assay test is the fact that the results may not be readily available and may take several days. In addition, the test must be performed prior to instituting any plasma therapy. Because TMA can cause rapid and progressive damage to vital organs relatively quickly, the luxury of waiting is not always the best approach [3, 13]. In the past, clinicians have heavily relied on plasma therapy for the management of TMA. However, with the advent of eculizumab, a distinction between TTP and aHUS is vital earlier on. Awaiting ADAMTS13 results in the context of aHUS may render the patient vulnerable to the unchecked devastation of aHUS. Plasma therapy (which is often the first intervention) prescribed to TMA patients would not provide targeted and optimal results in patients with aHUS [8].
Recent data have emphasized that surrogate markers for severe ADAMTS13 deficiency can facilitate prompt diagnosis and may serve as a guide to treatment in patients presenting with a TMA [14-17]. It has been documented that a vast majority of patients with severe ADAMTS13 deficiency demonstrate a serum creatinine level less than 2.26 mg/dL and a platelet count less than 30×109/L (Table 1). In addition, Coppo et al. [15] reported antinuclear antibodies in 53% of patients with thrombotic microangio- pathy associated with severe ADAMTS13 deficiency. A prediction model based on these three parameters indicates a specificity of 98.1% and a sensitivity of 46.9% for severe ADAMTS13 deficiency [15]. The sensitivity increases to 98.8% and the specificity decreases to 48.1% when only one criterion is present. Therefore, when thrombotic microangiopathy a associated with a serum creatinine level ≥ 2.26 mg/dL and a platelet count ≥ 30×109/L, TTP is a highly unlikely diagnosis and atypical HUS should be strongly considered (Table 1) [14-17]. This approach can ead to early diagnosis of atypical HUS in the proper context and timely initiation of complement inhibition therapy using a humanized monoclonal antibody (eculizumab).
Serum creatinine level, platelet count and antinuclear antibodies in thrombotic thrombocytopenic purpura.
| Variable | Ferrari et al. n=35 | Coppo et al.* n=160 | Vesely et al.* n=18 | Scully et al. n=80 |
|---|---|---|---|---|
| Serum creatinine, mean (mg/dL) | 1.26±0.59 | 1.29±0.77 | 1.8±1.2 | 1.1 (0.6-5.8) |
| Serum creatinine ≥ 2.26 mg/dL | 8.6% | N/A | 16.7% | N/A |
| Platelet count, mean (×109/L) | 17.4±13.7 | 17.4±14.2 | 11.6±6.9 | 13.5 (4-84) |
| Platelet count ≥ 30 (×109/L) | 11.4% | N/A | 0% | N/A |
| Antinuclear antibodies | N/A | 53% | N/A | N/A |
Data are expressed as mean ± standard deviation or percentage. * Patients with severe ADAMTS13 deficiency (less than 5% of normal activity). Numbers in parenthesis refer to the range of variable. N/A, not available.
In general, Low C3 and normal C4 concentrations in the serum indicates the activation of the alternative pathway of the complement system with an understanding that decreased serum C3 level is not specific for aHUS [3]. Furthermore, the demonstration of normal C3 and C4 concentrations does not exclude the diagnosis of aHUS [3]. Finally, not all of the individuals who have an abnormality of complement proteins develop aHUS [3, 8]. Therefore, the demonstration of complement protein abnormalities is not required to make the diagnosis of aHUS [3, 10].
It should be noted that clinical presentations of TTP and HUS have challenged traditional wisdom [1, 3, 18-20]. In this context, TTP was thought to predominantly have neurological involvement. Nevertheless, altered mental status, focal neurological deficits and seizure have been reported in approximately 10% of patients with aHUS [8]. Moreover, nearly half of the children with aHUS are reported to have neurological involvement [18]. The presence of diarrhea has been thought to indicate STEC- HUS. However, nearly one third of the patients with aHUS present with diarrhea [19]. Once considered a disease of children, aHUS is increasingly diagnosed in the adult population [21, 22]. Consequently, conventional methodo- logy of associating certain clinical features with TTP, STEC- HUS and aHUS is not an optimal approach.
It has been estimated that approximately half of the patients presenting with aHUS progress to end-stage renal disease within one year and a quarter of the patients die during the acute attack of the disease [1-4]. Even when transplanted, aHUS has a high rate of recurrence and subsequent damage to the transplanted organ and loss of graft [23-25]. Complement inhibition is emerging as an important strategy in stabilizing the platelet count, halting hemolysis and ameliorating renal injury [8, 13]. A humanized monoclonal antibody (eculizumab) has been successfully used for the management of aHUS [8]. In this context, establishing the diagnosis of aHUS in a timely fashion gains importance as eculizumab administration can directly limit the target organ damage. Prompt diagnosis is also important as another form of therapy (such as plasma exchange) is required for the treatment of TTP. At present, eculizumab is not indicated for the treatment of STEC-HUS.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work received no funding from public, commercial or not-for-profit organizations.

