
Continued Eculizumab Therapy for Persistent Atypical Hemolytic Uremic Syndrome
Authors Info & Affiliations
Abstract
Atypical hemolytic uremic syndrome (atypical HUS) is characterized by endothelial injury and microvascular thrombosis resulting in microangiopathic hemolytic anemia, thrombocytopenia, and ischemic injury to organs, especially the kidney. Activation of complements is involved in the pathogenesis of atypical HUS. Eculizumab, a neutralizing monoclonal antibody directed against complement C5, has shown salutary effects in patients with atypical HUS. In this report, we present a 23-year-old man with atypical HUSwho was treated with eculizumab. During the first four weeks of treatment, eculizumab failed to achieve a remission. Microangiopathic hemolytic anemia and thrombocytopenia persisted, while renal function deteriorated necessitating initiation of hemodialysis. Continuation of eculizumab therapy, however, led to marked improvement in hemolytic anemia, thrombocytopenia, and renal function. After 10 weeks of eculizumab therapy, hemodialysis was discontinued. At 5-month follow-up, serum creatinine was 1.1 mg/dL with continued eculizumab therapy every other week. In addition, platelet count was normal, while there was no evidence of hemolysis. We conclude that in patients with persistent atypical HUS continued treatment with eculizumab can be helpful in achieving remission.
INTRODUCTION
Hemolytic uremic syndrome (HUS) is characterized by microangiopathic hemolytic anemia, thrombocytopenia and ischemic injury to organs (especially the kidney) due to endothelial injury and microvascular thrombosis [1-3]. In a majority of patients with HUS, thrombotic microangiopathy is preceded within a week by a bacterial infection such as gastroenteritis, often hemorrhagic, due to shiga toxin-producing E. coli [1, 3]. In approximately 10% of patients with HUS, a bacterial infection is not identified (so-called atypical HUS) [1-4]. Accumulating evidence suggests a role for the complement system in the pathogenesis of atypical HUS [3-6]. In approximately half of patients with atypical HUS, hereditary defects in complement regulatory proteins result in activation of the complement system [5].
Administration of plasma (either in the form of plasma exchange or plasma infusion) has remained the standard treatment for atypical HUS for over three decades [1, 2, 5, 6]. While plasma infusion restores deficient regulatory proteins, plasma exchange has the additional benefit of removing autoantibodies that inhibit complement regulatory proteins. However, long-term treatment with this form of therapy can be difficult to achieve. Additionally, recent data have emphasized that patients with atypical HUS can fail to respond to aggressive plasma therapy [5, 7-9]. A neutralizing monoclonal antibody directed against complement C5, eculizumab has shown salutary effects in patients with atypical HUS by inhibiting complement-mediated thrombotic microangiopathy [5, 7-9]. The U.S. Food and Drug Administration has recently approved eculizumab for the treatment of atypical HUS [5].
In this article, we present a young man with atypical HUS presenting with microangiopathic hemolytic anemia, thrombocytopenia and renal failure. Because plasma exchange and glucocorticoids did not improve hematological and renal manifestations, eculizumab therapy was initiated. Although no sustained response was observed with eculizumab induction therapy, continued treatment resulted in reversal of renal failure and complete hematological remission. The present case suggests that continued eculizumab therapy might induce remission in persistent atypical HUS.
CASE REPORT
A 23-year-old Caucasian man with no significant past medical history was seen in an urgent care clinic with nausea, vomiting and upper abdominal pain. Physical examination was unremarkable. However, hematuria was noted on urinalysis and the patient was referred to the nearest emergency room. Laboratory tests revealed a hemoglobin level of 10.4 g/dL and a platelet count of 90,000/μL. Peripheral blood smear revealed numerous schistocytes. Serum lactate dehydrogenase was elevated, while serum haptoglobin was low. Serum creatinine and blood urea nitrogen levels were 8.1 mg/dL and 65 mg/dL, respectively. A diagnosis of thrombotic microangiopathy was made and the patient was admitted for further management.
At the time of admission the patient was in no acute distress. Apart from mild pallor, the remainder of the physical examination was unremarkable. The family history was notable for thrombotic microangiopathy in a first cousin who died at age 36 after receiving 3 kidney transplants. In addition, the father of this cousin suffered from renal failure of unknown cause and died at age 40.
Laboratory tests revealed hemolytic anemia, thrombocytopenia, azotemia, low complement C3 and normal complement C4. There were numerous schistocytes on blood films. Stool culture was negative for Shiga toxin-producing Escherichia coli, and serum ADAMTS13 activity was normal (>85%). Based on clinical grounds and biochemical abnormalities, the patient was diagnosed with atypical HUS.
The patient was started on daily plasmapheresis and prednisone 1 mg/kg/day. However, over the ensuing days the hemoglobin concentration and platelet count declined to 7.2 g/dL and 40,000/μL, respectively. In addition, renal function remained poor necessitating initiation of hemodialysis because of hyperkalemia and metabolic acidosis. Considering deterioration in renal function and hematologic profile despite plasma exchange and high-dose glucocorticoids, eculizumab therapy was started. To provide prophylaxis against meningococcal infections (a reported complication of eculizumab therapy) a meningococcal vaccine and penicillin were administered. The patient was induced with eculizumab 900 mg intravenously once a week for 4 weeks followed by maintenance therapy with eculizumab 1200 mg intravenously every 2 weeks (Fig. 1). During the first 4 weeks of eculizumab therapy, there was an improvement in platelet count and hemoglobin concentration following each dose. However, the effect was mild and transient (Fig. 1). Meanwhile, renal function remained poor requiring intermittent hemodialysis. The patient’s overall condition stabilized and he requested discharge from the hospital.
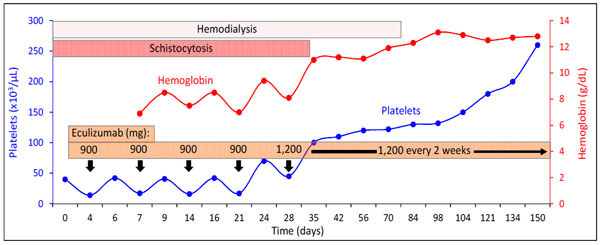
The impact of eculizumab therapy on platelet count and hemoglobin level.
Following discharge, the patient continued the eculizumab maintenance therapy (1200 mg every two weeks) and hemodialysis on an outpatient basis. After receiving hemodialysis for a total of 12 weeks, the patient reported increasing urine output. A 24-hour urine collection disclosed creatinine clearance of 36 ml/min. Therefore, hemodialysis was discontinued. Two weeks later, serum creatinine concentration was 2.4 mg/dL, and serum electrolytes, including bicarbonate, were within normal range. Hemoglobin concentration and platelet count were 12.2 g/dL and 180,000/μL, respectively. Serum lactate dehydrogenase and haptoglobin levels normalized. Mild hypertension was treated with lisinopril, which led to normalization of blood pressure. Hypercholesterolemia was treated with atorvastatin, which decreased LDL to 110 mg/dL and increased HDL to 55 mg/dL.
At five-month follow-up, the patient continued to do well on eculizumab 900 mg intravenously every 2 weeks. Serum creatinine declined to 1.1 mg/dL, platelet count was stable at 260,000/μL, and hemoglobin level was 12.8 g/dL. The patient was maintained on lisinopril, atorvastatin and ascorbic acid 500 mg/day.
DISCUSSION
Loirat [6], Schmidtko [2], Laurence [3] and Noris [7] have emphasized that thrombotic thrombocytopenic purpura (TTP), shiga toxin-producing Escherichia coli induced HUS (STEC-HUS) and atypical HUS cause microangiopathic hemolytic anemia, thrombocytopenia and target organ injury [1]. Indeed, from a clinical standpoint, the diagnosis of thrombotic microangiopathy relies on the demonstration of thrombocytopenia (platelet count <150,000/μL or a 25% decrease from baseline), microangiopathic hemolytic anemia (schistocytes, elevated LDH, decreased haptoglobin, decreased hemoglobin) and target organ injury (such as abnormal renal function tests, elevated liver function tests, neurological symptoms, gastrointestinal symptoms, increased pancreatic enzymes, etc.) [3]. Because endothelial injury is the major underlying pathology, clinically TTP, STEC-HUS and atypical HUS are often indistinguishable from one another with a concession that lungs are rarely involved in patients with TTP [1, 4]. In this context, laboratory tests often help a great deal in establishing the diagnosis [3]. While Coombs-negative hemolytic anemia (schistocytes), increased LDH, low haptoglobin, thrombocytopenia, and indicators of target injury (such as increased BUN/Cr, elevated liver function tests and pancreatic enzymes etc.) can be common to the three major TMAs, specific investigations such shiga toxin test and ADAMTS13 activity can be very helpful in distinguishing among the three entities. The current case demonstrated both normal ADAMTS13 activity and negative shiga toxin test, favoring the diagnosis of atypical HUS [3]. The current case also demonstrated low C3 and normal C4, indicating activation of the complements through the alternative pathway.
Recently, eculizumab therapy has been shown to be successful in the management of atypical HUS [5, 7-9]. In the case presented, eculizumab induction therapy did not produce immediate results. In fact, there were fluctuations in hemoglobin concentration and platelet count during eculizumab therapy. While there was an improvement in anemia and thrombocytopenia after eculizumab infusion, the effects were not sustained and the response was short lived. Indeed, there was a time during the induction phase of this patient where discontinuation of eculizumab therapy was contemplated due to the lack of a sustained positive impact. However, continued therapy with eculizumab not only eventually halted the thrombotic microangiopathy but also reversed the target organ injury. Platelet count and hemoglobin stabilized, hemodialysis was discontinued and serum creatinine ultimately declined to 1.1 mg/dL.
It is worth mentioning that specific mutations in the alternative pathway of the complement system can cause atypical HUS. These mutations include, but are not limited to, complement factor H, I, B (CFH, CFI, CFB) and membrane co-factor protein (MCP) [2, 6, 10]. In addition, neutralizing autoantibodies to complement regulatory proteins can also lead to activation of the complement system and cause atypical HUS. Specific mutations have been found in 50% of the patients with atypical HUS [2, 6, 10]. However, not all of the mutations have been identified. We did not investigate the presence of a genetic mutation in our patient. However, family history of a first cousin with the diagnosis of presumed TTP and multiple failed renal transplants points to this possibility. However, an investigation to assess a specific mutation may be helpful from prognostic as well as treatment standpoint (liver-kidney or kidney only transplant/replacement of a particular complement protein). In this context, a recombinant CFH is available and has produced positive results in an experimental setting [11].
Atypical HUS is a rare devastating disease with high morbidity and mortality. It can result in a 25% death rate during the acute phase and up to 50% of the patients can progress to end stage renal disease [1]. Recent studies have emphasized that eculizumab is helpful in stabilizing the platelet count, halting hemolysis and improving renal function [5, 7-9]. The current case demonstrated continued fluctuations with decline in platelet count and hemoglobin level after initial improvement with infusions during the first month. However, continued therapy resulted in stabilization and improvement in both platelet count and hemoglobin level and the patient was eventually taken off dialysis therapy. Based on the case presented, we suggest that initial less-than-optimal response to eculizumab therapy should not deter clinicians from using this form of therapy.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The project was not funded by any grants, funding agencies or pharmaceutical company. None of the authors have any conflict of interest.

