All published articles of this journal are available on ScienceDirect.

Acute Glomerular Diseases in Children
Abstract
Glomerulonephritis [GN] is one of the common acquired pediatric renal disorders encountered in clinical practice. The clinical manifestations include gross or microscopic hematuria, proteinuria, and nephrotic syndrome. Renal dysfunction and hypertension may also be present in many patients. Etiopathogenesis of GN can be idiopathic in a large majority, while some may result from infections or known immune disorders. Several of these disorders are now believed to arise from dysfunctions of podocytes and are grouped under the heading of “podocytopathies”. This review focuses on the clinical manifestations and management of the common forms of acute GN encountered in children.
INTRODUCTION
In the pre-renal biopsy era, most forms of nephritis and nephrotic syndrome were consdered as “Bright’s disease” [1]. With a better understanding of the pathology and pathogenesis of renal diseases, the term glomerulonephritis [GN] is used in the contemporary scientific literature to define non-infectious inflammatory lesion affecting the glomeruli. The clinical manifestations of GN encompass a spectrum that may include gross or microscopic hematuria, proteinuria, and nephrotic syndrome. Renal dysfunction and hypertension may also be present in many patients. Etiopathogenesis of GN also varies, being idiopathic in a large majority, while some may result from infections, such as hepatitis or human immunodeficiency virus-1 [HIV-V-1], or from known immune disorders, such as systemic lupus erythematosus [SLE].
CLASSIFICATION
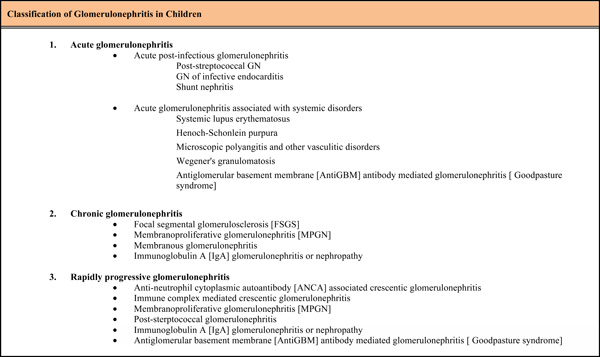
From a clinical perspective, GN can be classified as acute or chronic (Table 1). A sub-group of patients with acute GN designated as “rapidly progressive GN” [RPGN] have an aggressive clinical presentation, progressive decline of renal function occurring over days to weeks, and presence of crescents around the glomerular tufts in renal biopsy. Chronic GN, on the other hand, has an indolent clinical course, usually presenting with proteinuria, nephrotic syndrome, hypertension and variable degree of renal dysfunction. Renal pathology in chronic GN is variable. Of course, some disorders overlap in classification, since they can cause both acute and chronic GN. This review will discuss common forms of acute GN seen in children.
Classification of glomerulonephritis in children.
 |
Clinical features of acute glomerulonephritis associated with post-streptococcal disease, IgA nephropathy and Henoch-Schonlein pupura.
| Clinical Parameters | Acute Post-Streptococcal GN | IgA GN | HSP Nephritis |
|---|---|---|---|
| Preceding URI | Streptoccocal infection 10-14 days before onset of gross hematuria. Pyoderma associated APSGN: latent period may be up to 6 weeks | 24-72 hours before onset of hematuria [synpharyngetic]. URI is often viral in nature. | May have preceding streptococcal infection. Streptococcal antibodies positive in 25-30% |
| Duration of hematuria | 7-14 days | 2-14 days. Recurrent gross hematuria with URI. | Gross hematuria occurs in minority of patients. Microhematuria is more common |
| Microhematuria | Can persist up to 1 year | Can persist, but may disappear in between gross hematuria. | Microhematuria is common, usually resolves in few weeks. |
| Serum complement C3 | Low | Normal | Normal |
| Other Clinical associations | Hypertension Hyperkalemia Oliguria Mild azotemia | Acute kidney injury Nephrotic syndrome | Typical rash Joint pains Abdominal pain Risk for intessusception |
| Renal Biopsy finding | Sub-epithelial “humps” - electron dense deposits | Mesangial cell and matrix proliferation Mesangial IgA, IgG and C3 complement deposits | Mesangial cell and matrix proliferation Mesangial IgA, IgG and C3 complement deposits |
| Prognosis | Excellent | Long-term risk of CKD and hypertension. | Risk of CKD in only those with severe renal pathology. Most patients recover |
Abbreviations: CKD=chronic kidney disease; HSP=Henoch-Schonlein purpura; IgA= immunoglobulin A; IgG= immunoglobulin G.
Oxford classification of renal biopsy scores in IgA nephropathy. Based on and modified from :Working Group of the International IgA Nephropathy Network and the Renal Pathology SocietyThe Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int. 2009; 76: 534-45.
| Morphologic Findings | Definition | Score |
|---|---|---|
| Mesangial cellularity [M] Segmental sclerosis [S] &n Endocapillary hypercellularity [E] Tubulointerstitial atrophy/ Fibrosis [T] | < 50% mesangial tufts with ≥ 3 cells > 50% mesangial tufts with ≥ 3 cells Absentspan> Present Absent Present 0-25% cortical area 26-50% cortical area >50% cortical | M0 M1 S0 S1 E0 E1 T0 T1 T2T2 |
Original World Health Organization [WHO] classification of lupus nephritis. Modified from: Churg J, Sobin LH: Renal Disease: Classification and Atlas of Glomerular Disease, Tokyo, Igaku-Shoin, 1982.
| Pathologic Class | Renal Morphology |
|---|---|
| Class I | Normal glomeruli: but immune deposits are noted by immunofluorescence and electron microscopy |
| Class II | Mesangial alterations only a) No mesangial hypercellularity, but mesangial immune deposits are seen b) Mesangial matrix and cellular hypertrophy with immune deposits |
| Class III | Focal, proliferative glomerulonephritis: <50% glomeruli affected. |
| Class IV | Diffuse glomerulonephritis: >50% glomeruli affected. Severe mesangial, endocapillary, or mesangio-capillary proliferation and/or extensive subendothelial deposits] |
| Class V | Membranous glomerulonephritis |
| Class VI | Advanced glomerulosclerisis |
ACUTE POST-STREPTOCOCCAL GLOMERULONEPH-RITIS
Acute post-streptococcal GN [APSGN] is the most common form of GN encountered in children. WHO estimates that 472,000 patients are affected globally with APSGN each year, and results in 5,000 deaths annually [2]. Most of the mortality is related to complications in the acute phase of the disease.
Epidemiology
Although APSGN may occur at any age, it is more commonly encountered between the age of 2 and 15 years [3-5]. Occurrence of APSGN below the age of 2 years is rare [6].A declining trend in APSGN has been well documented in the United States from early1960s, especially in the impetigo associated disease [7]. In a recent analysis of APSGN, Ilyas noted that the prevalence of the disease had decreased from 2.18/100,000 population in the 1959-1973 cohort, to 0.64/100,000 in the 1999-2006 cohort [8]. ASPGN, however, remains a public health concern in developing countries, especially in the tropical regions [9]. APSGN follows upper respiratory or skin infection with group A β-hemolytic Streptococcus. Pharyngotonsillitis is a more common antecedent to APSGN in temperate climates and occurs during winter and spring months. Impetigo-associated APSGN, on the other hand, is more common during summer and autumn months and occurs more frequently in the tropical regions [7-9].
Clinical Manifestations
The onset of APSGN usually occurs 7-14 days after pharyngotonsillitis, but can be longer in the impetigo-associated disease [10]. Typical clinical features of APSGN consists of a combination of gross hematuria, oliguria, hypertension, mild lower extremity or periorbital edema, and a modest impairment in renal function (Fig. 1). Dull abdominal pain or flank pain and malaise may be present [3,4,10]. Mild fever can be seen in about half of the patients, often attributed to an underlying tonsillopharyngitis. Gross hematuria may last for a few weeks, but microscopic hematuria may persist for several months.
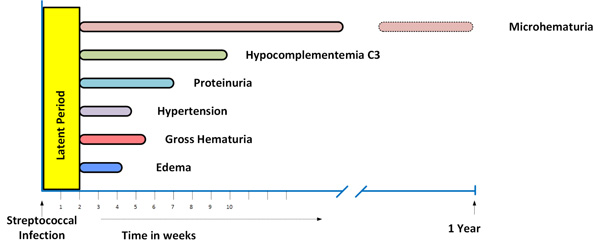
Time course of clinical and laboratory manifestations of post-streptococcal glomerulonephritis. Reproduced with permission from: Clinical Pediatric Nephrology, 3rd edition. Kher K, Schnaper HW, Greenbaum LW [Editors]. Taylor and Francis, Oxford, UK.
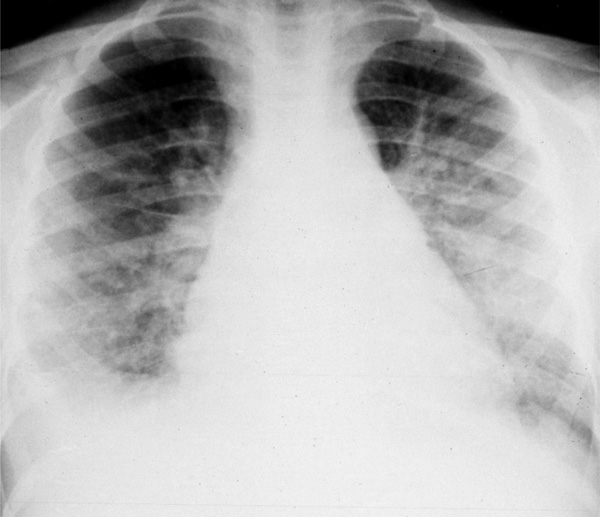
Chest X-ray of a patient with post-streptococcal glomerulonephritis showing pulmonary edema. He presented with difficulty in breathing, oliguria and “brownish urine”.
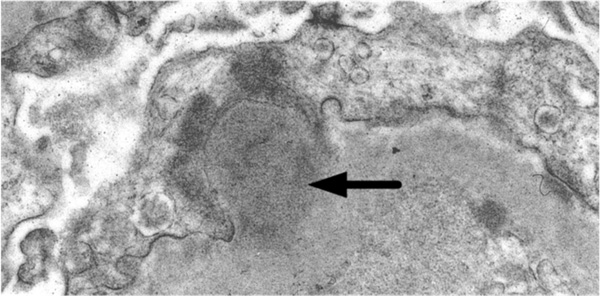
Electron microscopy of renal biopsy in a patient with post-sterptococcal glomerulonephritis. A characteristic large subepithelial electron dense deposit, or “hump” is seen [arrow].
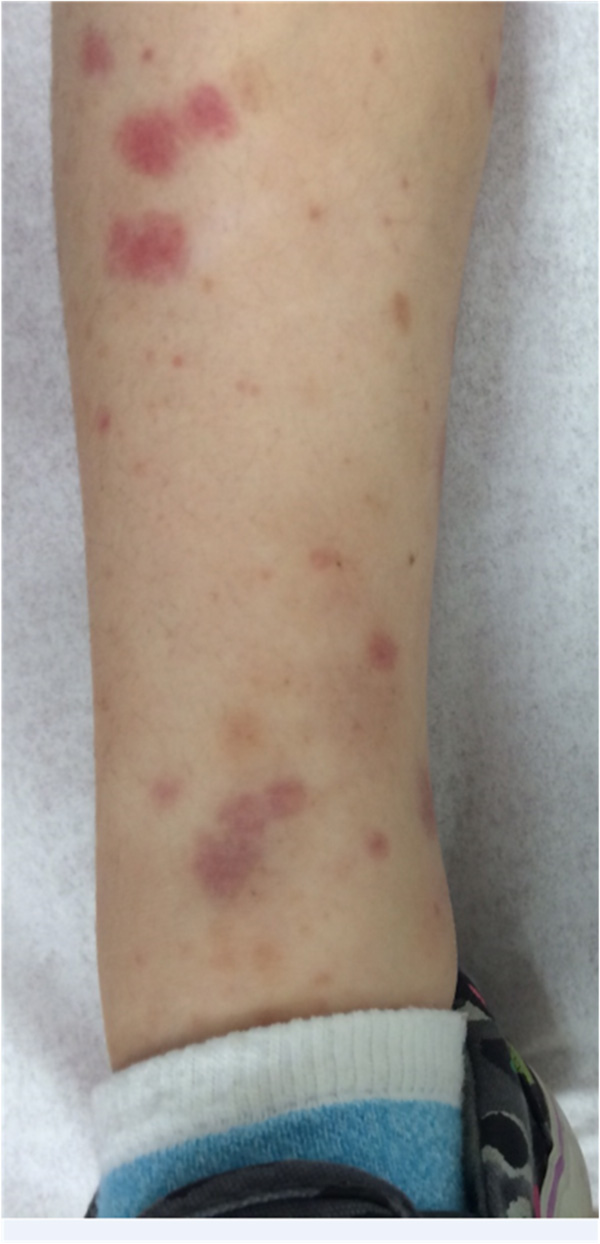
Characteristic rash in the lower extremities in a patient with Henoch-Schönlein purpura. Patient had mild nephritis, characterized by microscopic hematuria.
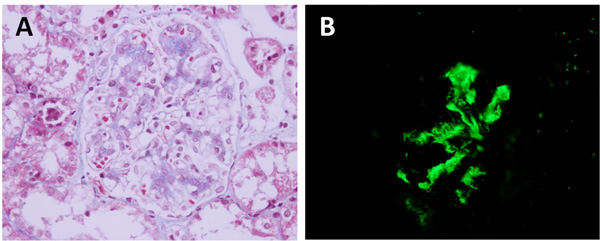
Renal biopsy findings in patients with Henoch Schölnlein purpura nephritis and IgA nephropathy. (A) Light microscopy showing moderate mesangial cell and matrix increase. Glomerular capillary loops are otherwise open. There is no evidence of extraglomerular crescents in this case. (B). Immunofluorescence staining with IgA antibody, showing IgA immune deposits in the mesangial region.
Hypertension is a common finding in patients with APSGN, occurring in over 80% of cases [4-6,11]. Severe hypertension, hypertensive encephalopathy and radiographically defined posterior reversible encephalopathy syndrome [PRES] have also been reported [12]. Mild pretibial and periorbital edema, caused by fluid retention and oliguria are noted in most patients. Severe fluid overload and pulmonary edema may be seen in some (Fig. 2). Asymptomatic APSGN may occur in contacts or family members of index cases [13].
Pathogenesis
In the past, Streptococcal M antigen has been considered as the primary nephritogenic antigen involved in the glomerular inflammatory response in APSGN [14].Available evidence, however, does not support the relevance of M antigen as a nephritogenic antigen [15]. Studies done in the last two decades have focused on two separate antigens, and possibly others which may be involved in the pathogenesis of APSGN. These antigens are: [1] nephritis-associated plasmin receptor NAPlr, and [2] streptococcal pyrogenic exotoxin B SPeB. Once deposited in the glomerulus, both of these antigens induce complement activation, and promote glomerular tissue injury, induce chemoattractants and inflammatory response in the mesangial and endocapillary sites [16-18].
Diagnosis
Apart from clinical features, the diagnosis of APSGN is established by : [1] demonstration of a rise of antibodies against streptococcal antigens, and [2] documenting a decrease in the serum complement C3. Two streptococcal antibodies that are commonly employed in the diagnosis are: a anti-streptolysin O ASO titer, and b anti-DNAse B antibody. ASO titer may not be significantly elevated in some patients early in the course of the disease and should be repeated to demonstrate a rising titer. ASO titers may remain low in patients with impetigo-associated APSGN. A decrease in serum complement C3 resulting from its consumption in APSGN is seen in the majority of patients early in the disease, and the decrease is profound in some patients. Complement C4 is usually normal but low levels have been noted in a minority of patients [19].
Renal dysfunction, characterized by elevated blood urea nitrogen BUN, and serum creatinine are seen transiently. Hyperkalemia is common in most patients and can be severe in some. A rising serum creatinine level and worsening of oliguria may be indicators of RPGN. These patients warrant a diagnostic renal biopsy.
Renal Biopsy
Renal biopsy is not necessary in most patients with APSGN. In patients with RPGN, the presence of significant proteinuria or non-resolving renal disease, a renal biopsy is an important diagnostic tool. The characteristic findings of APSGN on renal biopsy consist of cellular proliferation within the glomeruli, chiefly affecting the endothelial cells and mesangium. Polymorphonuclear infiltration within the inflamed glomeruli is also usually present. In cases with RPGN, extraglomerular proliferation and crescent formation are usually present. Immunofluorescence microscopy shows immune deposits consisting of immunoglobulin G IgG, along with complement C3. Electron-microscopy demonstrates presence of large sub-epithelial electron dense deposits or humps (Fig. 3).
Treatment
Most patients with APSGN respond well to conservative therapy consisting of: [1] fluid restriction, [2] sodium and potassium restriction, [3] treatment of fluid overload with diuretics, and [4] treatment of hypertension. Loop diuretics furosemide may also be helpful in decreasing serum potassium level in patients with mild hyperkalemia. Hyperkalemia may require treatment with potassium exchange resins sodium polystyrene sulfonate, Kayexalate®. Dialysis therapy may become necessary in patients with severe renal dysfunction, hyperkalemia or fluid overload, especially those with RPGN.
If group A beta hemolytic streptococci [GABS] are documented in the throat culture, or from skin lesions, the patient should be treated with appropriate antibiotic therapy. Although antibiotic therapy does not alter the clinical course of APSGN, elimination of nephritogenic group A beta streptococci as a reservoir of infection is important from an epidemiological perspective and preventing spread of the infection in the community. Antibiotic therapy has been found to be particularly helpful in curtailing the spread of GABS in epidemics.
Prognosis
Mortality of APSGN during the acute phase of the disease is well known and ~5000 deaths occur annually around the globe according to WHO estimates [2]. The mortality rate in developed countries has declined dramatically. Hadiwijayaet al. reported a mortality rate of 5.2% in Indonesia, while none of the 220 patients reported in a recent Japanese study died [20,21]. Mortality during the acute phase of the illness is usually due to complications, such as hyperkalemia, fluid overload, hypertensive emergencies and uremia.
Long-term prognosis of APSGN in children is excellent. Microscopic hematuria usually resolves in 3-6 months but may persist in a rare patient for as long as 4 years [21]. Proteinuria is usually transient in uncomplicated APSGN, and resolves in a 2-12 weeks in over 80% cases, and in 98% by the end of the first year [21]. Hypertension resolves in most patients within the first two weeks, and renal function normalizes around the same time.
HENOCH-SCHÖNLEIN PURPURA NEPHRITIS
Henoch-Schönleinpurpura HSP is a leukocytoclasticvasculitis characterized by rash, systemic manifestations and development of GN in some children. Most patients with GN present as an acute clinical disorder, although an occasional patient may develop chronic GN and nephrotic syndrome, with progressive decline in renal function.
Epidemiology
The incidence of HSPhas been estimated to be ~20.4 per 100,000 population per year [22].HSP primarily affects children younger than 10 years of age. Mean age at onset is ~6 years with slight male preponderance, but the disease has been reported in children as young as 6 months of age [23,24]. The disease is more common in the fall and spring [23-27]. Geographic clustering, or micro-epidemics have also been reported occasionally [28].
Pathogenesis
HSP and IgA nephropathy are now considered to be the two ends of the spectrum of the same disease. Some have gone so far as to characterize IgA nephropathy as HSP without rash [29]. This is based on the observation of similar renal pathology and immunofluorescence findings, as well as abnormalities in circulating serum IgA in both of these disorders. Pathogenesis of the HSP is discussed below in conjunction with IgA nephropathy.
Clinical Manifestations
Patients with HSP often present with history of a prodrome, usually an upper respiratory infection, followed by onset of abdominal pain, joint pain and swelling. Ankles and knees are commonly affected, but other large joints may also be affected. The child’s primary complaint may be difficulty in walking. A typical rash consisting of palpable purpuric lesions, usually begins around the medial malleolus and spread towards buttocks (Fig. 4) [23]. Extensor surfaces of the arms, particularly around elbows as well as the trunk are also affected, but the face is usually spared. In a rare patient, the onset of rash may be delayed by several weeks or months in relation to gross hematuria or proteinuria. Scrotal edema may be severe and may mimic testicular torsion [30]. Resolution of clinical symptoms usually occurs within two weeks, and is often complete by 4 weeks. Recurrence of rash is common and has been noted in 40-50% of cases, especially with renal involvement. Recurrences commonly occur in the first six months after the initial onset of the disease [23,31]. Less common manifestation include cerebritis, pancreatitis, and pulmonary hemorrhage [32-34].
Renal disease in HSP characterized by microscopic or gross hematuria, proteinuria and nephrotic syndrome, occurs in 30-40% cases [23,33]. Of these, hematuria is the most common feature. Mild proteinuria occurs in about 25% of unselected cases [23]. Severe proteinuria, nephrotic syndrome or nephritic-nephrotic manifestations [hematuria, proteinuria, renal dysfunction] occur in 5-10% of patients, and indicates severe renal disease [23,35]. These patients require close follow up and monitoring.
Diagnosis
European League Against Rheumatism/ Paediatric Rheumatology International Trials Organisation/ Paediatric Rheumatology European Society has proposed the following criteria for diagnosis of HSP with the mandatory presence of purpura or petechiae with lower limb predominance, plus one of four criteria: [1] abdominal pain; [2] skin biopsy demonstrating IgA; [3] arthritis or arthralgia; and [4] renal involvement manifesting as microscopic or gross hematuria and proteinuria [36]. BUN and serum creatinine elevation may be seen in those with significant nephritis. Patients with severe HSP nephritis may present with the nephrotic syndrome. Serum complement C3 is usually normal, but may be decreased in some patients [23,37]. Evidence of recent streptococcal infection may be seen in up to 20-30% cases [23].
The presence of IgA in the skin biopsy of patients with HSP is well known, and has been recognized as a diagnostic criterion of vasculitis in this disorder [36, 38]. Indeed, a skin biopsy rather than a renal biopsy has been considered as an adequate diagnostic test in patients with HSP nephritis [39]. In most cases of HSP nephritis, however, a renal biopsy is not necessary. Patients with progressive proteinuria or progressive decrease in renal function should be referred to a pediatric nephrologist, and a renal biopsy should be considered. A rapidly increasing serum creatinine in these patients may be indicative of crescentic or RPGN. An early renal biopsy and institution of therapy is an important key to renal outcome in such patients.
Treatment
Treatment of most patients with HSP is largely symptomatic and supportive. Non-steroidal anti-inflammatory drugs [NSAID] may be used for relief of joint pain. Use of oral corticosteroids has been shown to be effective in ameliorating the symptoms of abdominal pain in HSP [40]. The prednisone dose is usually 1-2 mg/kg /day for 1-2 weeks, and then tapered.
Evidence based data on management of HSP nephritis is scant. Corticosteroids have not been shown to be effective in preventing nephritis in double blind controlled trials [40].However, in a meta-analysis of published literature [1956 to January 2007] corticosteroids were noted to reduce the odds of developing persistent renal disease [41]. Mild nephritis, characterized by hematuria and sub nephrotic proteinuria usually resolves on its own and does not require intense corticosteroid treatment or immunotherapy [42]. Urinary findings usually abate in 4-6 weeks in these patients.
Numerous studies have documented an adverse outcome of severe renal disease in HSP. Those with nephritic-nephrotic clinical features, nephrotic syndrome, renal dysfunction, and hypertension at presentation are at an increased risk to develop chronic kidney disease [CKD ] and ESRD later on [43-48]. Treatment of this subset of children remains controversial. Data on the use and effectiveness of corticosteroids in these patients is unclear, since these have often been used in conjunction with other immunosuppressive agents [49].
Addition of cyclophosphamide to corticosteroids has been reported to be beneficial in patients with severe HSP nephritis [50]. In contrast, Tarshihet al. conducted a randomized controlled trial of 28 patients with histologically severe HSP nephritis who were treated with cyclophosphamide orally at a dose of 90mg/m2/day for 42 days [51]. Another 28 patients in the control group received only supportive care. Cyclophosphamide treatment did not improve the renal outcome of patients, as judged by incidence of CKD or ESRD.
Other immunosuppressive agents that have been used in the treatment of severe HSP nephritis include azathioprine and mycophenolatemofetil [MMF] [52-54]. The role of MMF in the treatment of HSP nephritis was encouraging in early studies [53,54]. Du et al. used MMF in a dose of 20 to 25 mg/kg per day in 12 children with severe HSP nephritis and nephrotic range proteinuria mean 5.6 grams per day for 10-15 months [53]. After a mean follow up of 3.9 years, all patients were free of proteinuria and all had normal renal function. Similarly, cyclosporine has also been useful in inducing remission of proteinuria in HSP nephritis and severe proteinuria [55]. Although effective, cyclosporine A dependence has been noted in some of these patients. Rituximab has been shown to be effective in inducing remission of patients with severe HSP nephritis who were refractory to other therapies [56]. Role of plasmapheresis in HSP nephritis is unclear. Some small studies have shown an improvement in patients with HSP and cerebritis, but others have no clear benefit in HSP associated RPGN [57,58].
Prognosis
The overall mortality of HSP nephritis is less than 1%, with most deaths occurring due to complications, such as hypertensive encephalopathy and systemic disease [35]. Although remission is common in many patients with HSP nephritis, residual renal disease can occur in a high proportion of children affected by this disease [35,45]. In 64 children with biopsy proven HSP nephritis, Schäreret al. demonstrated renal survival in 79%, but CKD, ESRD or death occurred in 21% of cases [35]. Of those with renal survival, 37% eventually had complete clinical remission, 30% had minor urinary abnormalities sub-nephrotic range proteinuria, no hypertension, normal renal function, and 12% had active renal disease high grade proteinuria, with/without hypertension. Hematuria alone, without associated proteinuria has been noted to have excellent long term outcome [39]. In a review of 1133 children with HSP, Narchi [48] noted that CKD never developed in patients with normal urinalysis but was seen in 1.6% of those with isolated urinary abnormalities, and in 19.5% of those who developed nephritic or nephrotic syndrome. The authors recommended follow up for 6 weeks after disease onset, since proteinuria can develop subsequent to the initial presentation.
Factors associated with poor renal outcome include: [1] high grade or nephrotic range proteinuria, [2] nephritic-nephrotic presentation proteinuria, hematuria, hypertension and renal dysfunction, [3] renal failure at initial presentation, and [4] severe renal biopsy changes, such as presence of crescents [35,44-46,59,60]. Interestingly, in a large Turkish study of 179 children who underwent renal biopsy for HSP nephritis, no correlation was found between presence of crescents in the renal biopsy and long term outcome [61]. Renal outcome of HSP nephritis in adults, especially in females, has been noted to be worse than in children and males [47]. Recurrence of HSP nephritis in renal transplants has been reported, sometimes leading to graft loss [62].
IGA NEPHROPATHY
IgA nephropathy IgAN, also known as Berger’s disease, was described as a distinct entity by Berger and Hinglais in 1968 in patients who had evidence of microscopic or gross hematuria as the dominant clinical manifestation [63]. IgAN now constitutes the commonest form of chronic GN in adults and accounts for about 50% of adults with CKD and ESRD world-wide [64, 65].
Epidemiology
The true incidence of IgAN is unclear. In the United States, IgAN accounts for ~10%, of all renal biopises [66]. In contrast, 20-35% of native kidney biopsies performed in Korea and Japan are diagnosed to have IgAN [67, 68]. South West Pediatric Nephrology group reported that 52% of children biopsied for gross hematuria carried the diagnosis of IgAN [69]. IgAN has been reported across all racial groups. One single center study reported that the incidence of IgANmay actually be higher among African American children in the United States [70]. Although not a familial disorder, large kindred of IgAN patients have been described in the United States and Italy [71, 72].
Clinical Manifestations
Patients with IgAN may present with dramatic symptoms of gross hematuria that follows an upper respiratory tract infection in the preceding 12-72 hours. This has often been labeled as “synpharyngetic hematuria” [73, 74]. Gross hematuria in APSGN, in contrast, occurs 10-14 days following a streptococcal URI. Gross hematuria usually lasts a few days, followed by microhematuria that can also vary in duration from few days to several weeks. Normal urinalysis may be seen in many patients with IgAGN. Proteinuria is usually seen early in the course of the disease. Nephrotic range proteinuria and nephrotic syndrome may be seen in patients with morphologically severe disease. Recurrent bouts of gross hematuria following URI are common in patients with IgAN, while persistent microhematuria may be a manifestation in about 50% of affected children. Clinical manifestations of IgAN in African American population conform to those seen in the Caucasian race [75].
A secondary form of IgAN has been described in patients with liver disease, such as cirrhosis [76]. Gastrointestinal disorders, such as celiac disease and dermatitis herpetiformis are also known to be associated with IgAN [77, 78]. IgAN has also been reported in association with viral infections, such as HIV, and hepatitis A, B and C infections [79-82].
Pathogenesis
The pathogenesis of IgAN and HSP is not entirely clear. A ‘four hit” model for pathogenesis of IgAN has been proposed [83-85]. The first hit is a genetic predisposition to produce an abnormal IgA1 molecule in both of these disorders. Circulating IgA1produced by B cells in response to respiratory infection in patients with IgAN and HSP is deficient in galactose in the hinge region of the heavy chains. The second hit consists of developing autoantibodies against the abnormal galactose deficient-IgA1. These antibodies can be either IgG or IgA. The third hit consists of deposition of IgA1-antibody complexes in the glomerular mesangium, a process that is less well defined. The mesangial immune complexes may be developed in-situ or result from deposition of circulating immune complexes. The fourth hit is the initiation of inflammatory pathways and local activation of complement within the glomeruli, and release of cytokines in the glomerular tissues. It is this final pathway that leads to glomerular injury and inflammation [83-86].
The pathogenesis of IgAN and HSP is believed to be similar, and the two disorders are believed to be the spectral ends of the same disorder. Some even go so far as to designate IgAN as “HSP without rash” [87, 88].
DIAGNOSIS
The diagnosis of IgAN requires a renal biopsy. However, during an initial discovery of microscopic hematuria, or first episode of gross hematuria, the diagnosis of APSGN needs to be ruled out (Table 2) by serum complement C3 level determination and evidence for GABS ASO titer, anti-DNAse B, throat culture. Renal function tests, quantification of proteinuria by a 24- hour urine collection or first morning urine protein/creatinine ratio should also be assessed. Elevated levels of abnormal galactose-deficient IgA1 levels are noted in 70-80% of patients with IgAN [89]. However, this test may not be available commercially in most clinical laboratories.
Other conditions that present with gross hematuria, and need to be considered in the differential diagnosis include Alport syndrome, thin-basement membrane nephropathy TBMN and membranoproliferative GN MPGN. Systemic lupus nephritis also presents with gross or microscopic hematuria and proteinuria, and should be ruled out by appropriate laboratory evaluation. All of the above mentioned clinical conditions also need a renal biopsy for confirmation of the clinical diagnosis.
Renal Biopsy
Renal biopsy findings in IgAN and HSP are similar; both disorders showing a variably increased mesangial matrix and cellularity on light microscopy. Immunofluorescence microscopy (Fig. 5) demonstrates a dominant presence of immunoglobulin A IgA, along with complement C3 and IgG [74, 90].
The International IgA Nephropathy Network and the Renal Pathology Society has proposed Oxford classification of IgAN or Oxford -MEST classification [91, 92]. This renal biopsy classification utilizes the degree of mesangial cellularity, segmental glomerulosclerosis, endocapillaryhypercellularity and tubulointerstitial fibrosis as the parameters of scoring (Table 3). The severity of renal biopsy is scored on a progressive scale of changes in each category, and the final score is indicated by a composite of each aspect of evaluation parameter. Although extraglomerular proliferation crescent formation is generally regarded as an indicator of poorer outcome in patients with GN, the Oxford classification of IgAN did not consider it to be an important variable in their study. In general, more severe morphologic disease correlates with a worse long-term outcome.
Treatment
There is no clear consensus about treatment of IgAN. Since proteinuria is considered a marker of progression of renal disease in a variety of disorders, including IgAN, treatment with angiotensin converting enzyme inhibitors ACEI should be considered in IgAN patients with significant or advancing proteinuria. Both ACEI and angiotensin receptor blockers ARB have been found to be useful in ameliorating proteinuria in children with IgAN [93, 94].
Corticosteroids alone may help patients with moderately severe disease. However, randomized controlled trials using corticosteroids alone in IgAN are limited. The South West Pediatric Nephrology group studied the effects of daily corticosteroid followed by alternate day therapy for a total of 2 years in children and young adults with IgAN. This study did not demonstrate any superiority of treatment with corticosteroids over placebo in preventing decline of GFR [95]. A recent meta-analysis of published studies of IgA in adults and children demonstrated that corticosteroid use in IgAN was associated with a lower risk of renal disease and proteinuria in the follow up periods [96]. Further subgroup analysis of the data demonstrated that long term, low dose use of steroids did not reduce the risk of renal disease. On the other hand, high dose oral or intravenous costicosteroids used for a short time was helpful in reducing the long-term risk of renal disease in this analysis.
The role of MMF in the treatment of IgAN is unclear. Several studies have demonstrated failure of a clear benefit of MMF in the treatment of IgAN of varying severities [97,98]. Transient and partial remission of proteinuria has, however, been noted by some [98]. A metanalysis of MMF used in IgA nephropathy also did not demonstrate any benefit in the treatment of proteinuria or protecting decline in renal function [99]. In children with IgAN resistant to steroid therapy, combining corticosteroid treatment with MMF has been noted to be beneficial in inducing remission of proteinuria in 63% patients [100].
Omega3 fatty supplements fish oil had been thought to be of benefit in ameliorating proteinuria in patients with IgAN [95]. The anti-proteinuric effect of omega-3 fatty acid therapy may be dose dependent [101]. A meta analysis of published data also suggested that despite ameliorating proteinuria, omega-3 fatty acids did not demonstrate any benefit in preventing a decline in renal function [102].
Combining corticosteroids with other immunosupres-sives, such as cyclophosphamide or azathioprine, alone or with warfarin and dipyridamole has been attempted quadruple therapy. In one such randomized controlled trial, azathioprine showed improvement in renal survival [103]. Because of the high risk for CKD in patients with severe renal pathology in patients with IgAN, most clinicians tend to use a combination of intravenous corticosteroids, and immunosupressives azathioprine, MMF, cyclosporine.
Prognosis
IgAN has a better outcome in children than in adults. Japanese experience with IgAN suggests that spontaneous remission of hematuria is possible in ~50% of children with the disease [104,105].Time to remission of hematuria and proteinuria can be several years [5-s], and recurrence of symptoms after prolonged remissions is well known [105]. Persistent microscopic hematuria or recurrent gross hematuria occurs in 35% cases and widely spaced or isolated episodes of hematuria are present in 10-15% cases [73-75]. Frequency of gross hematuria or degree of microscopic hematuria do not predict long term prognosis [106]. On the other hand, the presence of proteinuria and its severity are well known to predict renal outcome in IgAN [107,108]. Evolution of ESRD, often as adults, is seen in ~20-25% pediatric IgAN cases [73-75,104,109]. Finnish data suggests that ESRD occur in 11% cases, while 39% were being treated for hypertension and proteinuria after a mean follow up of 18.7 years range: 8.5-29.8 years [109]. Available data in adults also suggests that renal outcome is worse in those with the nephrotic syndrome [110].The severity of histological changes on renal biopsy at onset, especially using the Oxford classification has also been shown to be predictive of outcome [111,112].
LUPUS NEPHRITIS
Systemic lupus erythematosus SLE is an autoimmune disorder that is associated with GN and has a high potential for CKD and ESRD. The clinical manifestations of SLE as well as lupus nephritis LN in children are highly variable and can simulate both acute and chronic GN at its onset. LN often needs to be considered in the differential diagnosis of acute nephritic syndrome and nephritic-nephrotic clinical presentation.
Epidemiology
LN can be either the presenting manifestation, or becomes evident at initial discovery of the disease in 60-80% of children [113-115]. In general, SLE as well as LN are more common in females. The disease is reported to be more prevalent in Asian and African American girls, as compared to Caucasians [116]. American College of Rheumatology ACR criteria for diagnosis of LN are: [1] persistent proteinuria defined as 0.5 g per day in a 24 hour urine collection or first morning spot urine protein/creatinine ratio of 0.5 nephritic urine sediment consisting of red blood cells RBCs, granular, tubular, or mixed casts. [3] Serologic diagnostic criteria of SLE, and [4] suggestive renal biopsy, when available [117]. Presence of renal failure is not included as a diagnostic criterion of LN in the current ACR definition.
Pathogenesis
SLE is characterized by a disordered immune system, wherein auto-antibodies are produced against numerous “self” antigens, such as native DNA, cytoplasmic proteins and nuclear proteins. The auto-antibodies, in turn, form circulating and in-situ immune complexes that result in inflammatory response in the affected tissues and organs. Within the kidney, immune complex deposition, and subsequent inflammatory response can affect the glomeruli, tubulointerstitial space, as well as the vascular structures. Variable involvement of these tissue compartments by the inflammatory response and damage is common in patients with LN.
Renal Pathology
The type of morphologic lesions encountered in LN is also diverse, ranging from almost normal glomeruli with mild mesangial proliferation to severe and diffusely proliferative GN. World Health Organization WHO provided a clinically relevant classification system for LN (Table 4) that has now been modified and enhanced by the International Society of Nephrology and the Renal Pathology Society [118].
Immunofluorescence microscopy in LN usually demonstrates presence of immunoglobulinsIgG, IgA and IgM, along with complement components, often referred to as “ full-house” immune deposition. Apart from the above noted lesions, renal biopsy may also show tubulointerstitial inflammation and fibrosis, presence of variable degrees of extra-glomerular cellular crescents and reticular structures
Clinical Manifestations
Manifestations of LN are varied, and can be microscopic or gross hematuria, along with proteinuria. Nephritic-nephrotic clinical features, with edema, hematuria, acute renal injury and hypertension are also commonly encountered. Patients with class V Membranous variant form of LN have heavy proteinuria and nephrotic syndrome as a dominant clinical finding. On the other hand, class III diffuse proliferative may have a nephritic-nephrotic presentation with hematuria-proteinuria, hypertension and elevated serum creatinine level. In an Italian registry of SLE nephritis, nephrotic syndrome, hypertension, and renal failure were present at onset in 40%, 28%, and 32%, respectively [119].
Diagnosis
Apart from studies to establish the diagnosis of SLE by various antibody titers and inflammatory markers, renal biopsy is essential in establishing the diagnosis of LN. Clinical features, urinary protein extcetion and urinary sediment evaluation may not provide sufficient evidence to diagnose LN in many patients, and “silent” LN has been well documented [120].
Treatment
Treatment of LN is largely dictated by the type of pathology seen on renal biopsy and the severity of the disease. Class I and II LN, which represent mild form of nephritis, is usually treated with corticosteroids to control of systemic disease. On the other hand, class III focal proliferative LN and class IV diffuse proliferative LN can be quite aggressive in their clinical course and carry a potential of adverse long term renal outcome, such as CKD and ESRD. For this reason, children affected by these classes of renal pathology need aggressive immunosuppression. Although numerous immunosuppressive options are now available for treatment of class III and IV LN, inravenous cyclophosphamide and MMF have emerged as the two primary induction therapies in children in a recent consensus statement [121].
Intravenous cyclophosphamide induction consists of administering a total of six monthly doses, initiated at 500 mg/m2 body surface area. Subsequent doses can be increased but not to exceed a maximum monthly dose of 1,500 mg [121]. MMF induction is dosed at 600 mg/m2/dose twice daily, with a maximum dose of 1,500 mg taken two times per day. Mycophenolate sodium MPA; Myfortic® may be used as an alternative to MMF at a dose of 400 mg/m2/dose twice daily and maximum dose of 1,080 mg twice daily. It is important to monitor and prevent side effects, especially bone marrow suppression and infection risks associated with the immunosuppressive therapies [121,122].
Immunosuppressive drug dose should be adjusted to the nadir white blood cell count. Extra-renal manifestations and the activity level of the disease should also be monitored during induction therapy. Glucocorticosteroids are often prescribed at modest doses during induction of LN. Total duration of induction regimens lasts 6 months, after which the patients are gradually returned to a maintenance schedule of either corticosteroids alone, or in combination with MMF.
The treatment of choice for class V membranous nephritis LN is with calcineurin inhibitor agents, such as cyclosporine, since they respond less favorably to corticosteroids and cyclophosphamide. Austin et al. demonstrated that at 1 year, the cumulative probability of remission was 27% with prednisone, 60% with intravenous cyclophosphamide and 83% with cyclosporine [123]. Data on the use of tacrolimus in LN is scant.
Prognosis
A decreasing trend of mortality of patients hospitalized with LN has been noted between the years of 2000-2009, and stands at 0.6% [124]. Progression of LN to CKD and ESRD is well documented. LN accounts for 5.8-6.4% of pediatric ESRD patients in the United States [125]. Renal survival was noted to be 44.4% at 5 years and 29% at 10 years between 1965 and 1992 [126].Poorer renal outcome and progression to ESRD is often associated with class III and IV proliferative nephritis LN [126, 127].
CONCLUDING REMARKS
GN is one of the common acquired pediatric renal disorders encountered in clinical practice. Resurgence of post-streptococcal GN since 1990s has made this to be the most common type of glomerulonephritis encountered, globally. Most chronic types of glomerulonephritidies manifest as nephrotic syndrome, or with a mixed nephritic-nephrotic clinical picture. A better understanding of some forms of chronic types of GN is emerging with a more clear understanding of the physiology of podocytes. Several of these disorders are now believed to arise from dysfunctions of podocytes and are beginning to be grouped under the heading of “podocytopathies”. Other developments in this area relate to development of newer drugs that target complement dysfunction in clinical conditions, such as membranoproliferative GN. Genetic studies are also bound to provide another crucial link to the pathogenesis of GN in the future.
ABBREVIATIONS
CONFLICT OF INTEREST
The author confirms that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

