All published articles of this journal are available on ScienceDirect.

Isolation of Glomerular Podocytes by Cationic Colloidal Silica-coated Ferromagnetic Nanoparticles
Abstract
Background:
Podocyte homeostasis plays a crucial role for the maintenance of physiological glomerular function and podocyte injury is regarded as a major determinant of development and progression of renal disease.
Objective:
Investigation of podocytes requires appropriate methods for their isolation. Previously reported methods use podocyte specific antibodies or transgenic mice with podocyte specific expression of fluorescent markers for isolation of podocytes by magnetic or fluorescence activated cell sorting.
Method:
Here, a novel, antibody-free method for isolation of podocyte protein and RNA from mouse glomeruli is described. Preparations of isolated glomeruli were added to a suspension of cationic silica-coated colloidal ferromagnetic nanoparticles. The nanoparticles bound to the negatively charged cell surfaces of podocytes residing on the outer surface of the isolated glomeruli. After enzymatic and mechanical dissociation of glomerular cells, nanoparticle-coated podocytes were isolated in a magnetic field. The method was tested in adult wild-type mice without renal lesions and in mice of two nephropathy models (Growth hormone (GH)-transgenic mice and transgenic mice expressing a dominant negative receptor for the glucose dependent insulinotropic polypeptide, GIPRdn) displaying albuminuria, glomerular hypertrophy and evidence for a reduced negative cell surface charge of podocytes.
Results:
The isolated cells displayed typical morphological and ultrastructural properties of podocytes. On average, 182,000 ± 37,000 cells were counted in the podocyte isolates harvested from ~10,000-12,000 glomeruli per mouse. On the average, the purity of podocyte isolates of these mice accounted for ~63 ± 18 % and the podocyte isolates displayed high mRNA and protein expression abundances of podocyte markers (nephrin and WT1), whereas the expression of endothelial (Cd31) and mesangial markers (Serpinb7) was significantly decreased in podocyte isolates, as compared to samples of isolated glomeruli. The numbers of cells isolated from GH- transgenic and GIPRdn-transgenic mice were not markedly different from that of wild-type mice.
Conclusion:
The described method represents an alternative for podocyte isolation, particularly in experiments where podocyte specific antibodies or transgenic animals with podocyte specific expression of fluorescent markers are not applicable.
1. INTRODUCTION
Glomerular visceral epithelial cells (podocytes) are highly specialized cells that cover the glomerular basement membrane (GBM) with interdigitating foot processes, between which the glomerular slit diaphragm is formed as the most selective part of the glomerular filtration barrier [1]. During the past decades, the crucial role of podocytes for maintenance of the physiological glomerular structure and function was recognized, and podocyte damage has been identified as a key determinant in development and progression of a variety of different renal disease entities [1, 2]. Investigation of podocyte specific mRNA- or protein expression patterns in distinct phases of physiological nephron development, or in defined stages of pathological glomerular alteration by transcript profiling- or proteome analyses, is regarded as a pivotal chance to facilitate an improved understanding of podocyte biology and of the molecular mechanisms involved in establishment of podocyte injury in vivo [1]. In analyses of gene- or protein expression data from samples of total kidney tissue or isolated glomeruli, expression signatures of podocyte origin might be overseen or misinterpreted, as they can become overlaid or distorted by signals derived from other kidney cell types [3, 4]. As well, in vitro analyses of primary or immortalized podocyte cell cultures harbour limitations, as cultured podocytes cannot completely mimic the complex characteristics of podocytes in vivo, and an authentic reproduction of cell culture conditions representative for comparable states of pathological podocyte alterations in vivo is doubtful [1]. Therefore, performance of podocyte-specific gene- or protein expression analyses necessarily requires appropriate techniques for generation of fresh podocyte isolates. Schroeppel et al. (1998) described a first method for RT- PCR based mRNA analysis of podocytes in vivo, using a micropipette to selectively aspirate single podocytes from microdissected glomeruli under visual control [5]. Apart from this single cell approach, only two methods for isolation of substantial numbers of podocytes for subsequent molecular analyses have so far been reported by Takemoto et al. (2006) [6], Murakami et al. (2010) [7], Boerries et al. (2013) [8], and by Brunskill et al. (2011, 2015) [9, 10], who used magnetic or fluorescence activated cell sorting for isolation of podocytes from murine kidney tissue. These very effective methods depend either on large quantities of podocyte-specific antibodies [7], or need to be performed on samples generated from transgenic animals with podocyte specific expression of fluorescent proteins, such as green fluorescent protein (GFP) [6, 8, 9].
The present study describes a novel, antibody-free, cost-efficient method for in vivo isolation of podocytes from mouse glomeruli, which is not depending on transgenic mice with podocyte specific GFP transgene expression. The basic principle of the method is schematically outlined in Fig. (1). Isolated glomeruli are incubated with a suspension of cationic colloidal silica-coated ferromagnetic nanoparticles (NPs) that bind to the negatively charged cell surfaces of podocytes on peripheral glomerular capillary loops. NP-coated podocytes are separated from other glomerular cells by enzymatic digestion and mechanical dissociation, and then isolated in a magnetic field.
2. MATERIALS AND METHODS
2.1. Chemicals, Reagents and Materials
2.1.1. Glomerulus and Podocyte Isolation
DynabeadsTM M-450 Epoxy (Art.: 1011) and Hanks balanced salt solution (HBSS, Art.: 14060-040) were purchased from Invitrogen; Collagenase A (Art.: 10103578001) and Collagenase/Dispase (Art.: 10269638001) from Roche, and the BDI MagnetTM Cell Separation Magnet (Art.: 552311) from Becton Dickinson. Trypsin (Art.: T4799), 0.2% Trypsin–EDTA (Art.: T4049), heparin (Art.: H3393), DNase I (Art.: DN25), and soybean trypsin inhibitor (Art.: T9003) were bought from Sigma-Aldrich, Germany. Cell strainers of 40 µm (Art.: 352340) and 100 µm (Art.: 352360) mesh width were purchased from Schubert & Weiss GmbH (Germany). Monofilament meshs with 1 µm and 25 µm mesh width (PA-1/115, Art-N°: SG00000001; PA-25/102; Art-N°: SG00000005) were bought from Schwegmann Filtrations-Technik GmbH (Germany).
2.1.2. Nanoparticle Synthesis
Iron(III)chloride (FeCl3, Art.: 157740), Iron(II) sulphate hydrate (FeSO4xH2O, Art.: 12353), 35 % Poly(acrylic) acid solution MG ~100 000 (PAA, Art.: 523925), 4-Morpholineethanesulfonic acid (MES, Art.: M3671), D-Sorbitol (Art.: W302902), 3-Aminopropyltriethoxysilane (APTES, Art.: A3648), and ammonia solution (NH3, Art.: 392685) were bought from Sigma-Aldrich.
2.1.3. Buffers and Embedding Media
Phosphate buffered saline (PBS, adjusted to pH 7.4: 8.00 g NaCl, 0.20 g KCl, 1.44 g Na2HPO4, 0.20g KH2PO4/l H2O); Coating buffer (CB adjusted to pH 7.4: 3.90 g MES, 8.77 g NaCl, 145.74 g Sorbitol/l H2O); Protein extraction buffer (PEB adjusted to pH 7.0: 10 mm Na2HPO4 at pH 7.0; 0.2% (wt/vol) sodium dodecyl sulfate solution; 10% (vol/vol) glycerine); Stripping buffer (SB: 37.5 ml of 250 mM TRIS buffer at pH 6.7; 30 ml of 10%; 82.5 ml H2O; 1050 µl ß-mercaptoethanol). Enzymatic solution for digestion of isolated NP coated glomeruli (0.2% trypsin–EDTA, 100 mg/ml heparin and 100 U/ml DNase I). Hanks’ Balanced Salt solution (HBSS) was obtained from Sigma-Aldrich, Germany. Epon (glycid ether 100) was purchased from Serva, Germany.
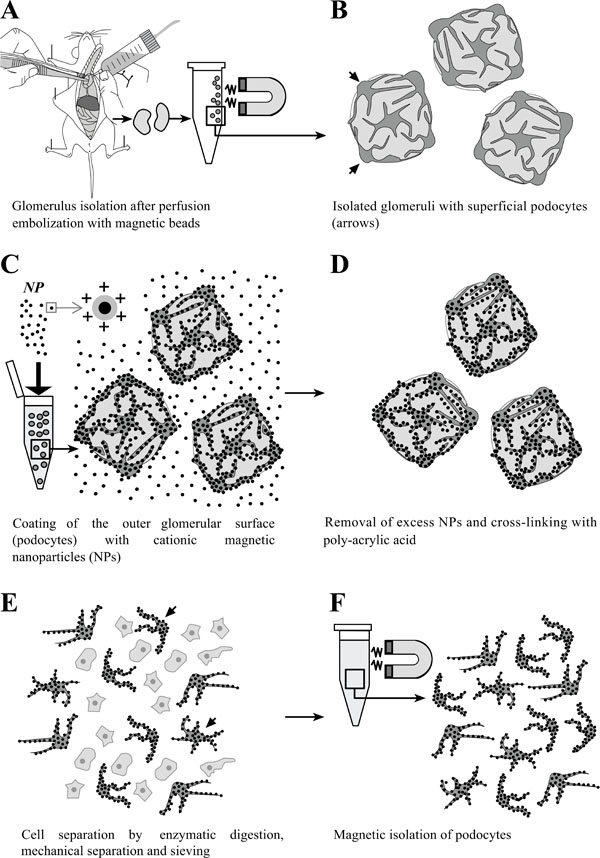
Schematic illustration of the principle of isolation of podocytes coated with cationic colloidal silica-coated ferromagnetic nanoparticles (NPs). A: Glomeruli are isolated after perfusion with spherical superparamagnetic beads. B: Simplified schematic illustration of isolated glomeruli. Podocytes residing on the outer glomerular surface are indicated by arrows. C: Incubation of isolated glomeruli with NPs, binding to the exposed negatively charged podocyte cell surfaces. D: Excess NPs are removed from the sample by sieving. Podocyte-bound NPs are cross-linked by addition of poly-acrylic acid, simultaneously neutralizing the exposed surfaces of NPs to avoid binding to other cell types after cell separation. E: Glomerular cells are dissociated by enzymatic digestion, gentle mechanic squeezing in a Dounce tissue grinder, and subsequent sieving. F: Isolation and repeated washing of NP-coated podocytes in a magnetic field.
2.1.4. Western-blot Experiments
A molecular weight standard (PageRuler) was purchased from Thermo Fisher Scientific, Germany. Blotting membranes (Immun-Blot® PVDF Membrane) were bought from Bio-Rad, Germany. Primary and secondary antibodies: Anti-mouse CD31 (PECAM-1) rat monoclonal antibody (Clone SZ31, Histonova, Germany), diluted 1:5000; Anti-Mouse nephrin polyclonal goat antibody (Cat. AF3195, R&D Systems, Germany), diluted 1:2000; Anti-GAPDH (14C10) rabbit monoclonal antibody (Cat. 2118L, CellSignaling Technology, USA), diluted 1:2000; Anti WT1 rabbit polyclonal antibody (SAB2104329, Sigma- Aldrich, Germany), diluted 1:900; Peroxidase conjugated goat anti-rabbit HRP-linked whole Ab from donkey (Cat. NA934V, GE Healthcare Life Sciences, USA), diluted 1:2000; Donkey anti-goat IgG HRP coupled antibody (LifeSpan BioSciences, Biozol Diagnostica Vertrieb GmbH, Germany), diluted 1:5000; Polyclonal rabbit anti-rat immunoglobulins/HRP (Code No. P045, Dako, Denmark), diluted 1:1000. ECL reagent (RPN2106) was bought from GE Healthcare Amersham Biosciences, Germany.
2.1.5. Quantitative RT-Real Time PCR (RT-qPCR) Analyses
The RNeasy-Mini kit was purchased from Qiagen, Germany. The high capacity RNA-to-cDNA kit and the power SYBR green PCR master mix were bought from applied biosystems, Germany.
2.1.6. Immunofluorescence-Cytochemistry and -Histochemistry
Fetal bovine serum was purchased from Thermo Fisher Scientific (Germany). Primary and secondary antibodies for immunofluorescence: Guinea pig polyclonal nephrin antibody (GP-N2, Progen Biotechnik, Germany), diluted 1:50; rat monoclonal anti-mouse CD31 (PECAM-1) antibody (#DIA310, Dianova, Germany), diluted 1:20; Donkey-anti-guinea pig IgG (H+L)-Cy5 (#706-175-148, Dianova, Germany), diluted 1:500; and Donkey anti-Rat IgG (H+L) (Clone: pAb)-FITC (#712-096-150,Dianova, Germany), diluted 1:200. 0.5% Triton X-100 was bought from Sigma-Aldrich, and Vectashield antifade solution from Biozol Diagnostica Vertrieb GMBH, Germany.
2.1.7. Immunohistochemistry
Primary and secondary antibodies used for immunohistochemistry: Rabbit polyclonal anti WT1 antibody (Sc- 192, Santa Cruz Biotechnologie, Germany), diluted 1:100; Rabbit polyclonal anti Podocalyxin antibody (Bs- 1345R, Acris Antibodies GmbH, Germany), diluted 1:100; Polyclonal Swine Anti-Rabbit Immonoglobulins/Biotinylated (E0353, Dako, Denmark), diluted 1:200; Polyclonal Goat Anti-Rabbit Immonoglobulins/HRP (P0448, Dako, Denmark), diluted 1:100. The Vectastain® ABC-Elite staining kit (Standard: HRP, Pk-6100) was purchased from Linaris Biologische Produkte GmbH, Germany.
2.1.8. Detection of Cationic Sites on Podocyte Cell Surfaces
Low molecular weight (1.8 kD) polyethyleneimine (PEI, Art. 408700) was bought from Sigma-Aldrich Chemie GmbH, Germany.
2.2. Synthesis of Cationic Colloidal Silica-Coated Ferromagnetic Nanoparticles
The details of the protocol for synthesis of cationic colloidal silica-coated ferromagnetic NPs (Figs. 2-4) presented below were determined empirically in a series of preliminary experiments. Magnetite (Fe3O4) nanoparticles (NPs) were prepared under a nitrogen (N2) protective atmosphere by chemical co-precipitation from an aqueous solution containing Fe3+/Fe3+ ions in a molar ratio of 2:1 upon addition of ammonia solution [12]. 1.52 g FeSO4 and 3.24 g FeCl3 were successively dissolved in 150 ml of completely deoxygenated water by vigorous stirring for at least 10 minutes at room temperature (RT). If necessary, the FeSO4 solution was heated until complete dissolution and then cooled down to RT before addition of FeCl3. Then, 40 ml of 25% aqueous ammonia solution were rapidly injected into the solution, whereupon a black-to-dark-brown precipitate formed. While the solution was further stirred, additional ammonia was added, until the pH reached 9-10. The magnetite was then washed by magnetic separation, performing four washes with deoxygenated water, two washes with 50% ethanol, and two washes with 100% ethanol. Subsequently, the washed magnetite was dried into a powder under vacuum at RT. Approximately 400 mg of dried Fe3O4 were sonicated (Branson Sonifier Cell Disruptor B 15, Emerson, Germany) in 15 ml of 50% ethanol for 25 minutes at output stage 7, to achieve a uniform dispersion of NPs. Then 1.6 g of APTES were added to the mixture under N2 atmosphere and stirred for 3 hours at 40°C. The APTES-modified Fe3O4 NPs were collected with a magnet, washed twice with 50% ethanol, and three times with deoxygenated water. The NP preparation was dried under vacuum and stored until further use under a N2 atmosphere for up to six weeks at 8°C, or at -80°C for longer storage periods. For podocyte isolation, suspensions of NPs in CB (10 mg/ml) were prepared and rigorously vortexed for one minute prior to use.
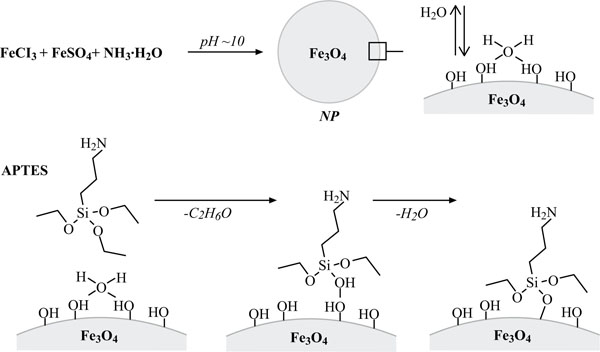
Synthesis of cationic colloidal silica-coated ferromagnetic nanoparticles (NPs). Top: Fe3O4 (magnetit) nanoparticle synthesis. Bottom: 3-Aminopropyltriethoxysilane (APTES)-hydrolysis and condensation at hydrated magnetit surface resulting in a positively charged NP surface (modified after Howarter and Youngblood, 2006) [11].

Cationic colloidal silica-coated ferromagnetic nanoparticles (NPs). A: NPs in solution. B: NPs demonstrate ferromagnetic properties in the field of a strong permanent magnet. C: Transmission electron microscopy of NPs. Bar = 100nm. Inset to C: Dried NP-powder.
2.3. Mice and Experimental Groups
All animal experiments were carried out in accordance with the German Animal Protection Law. The details of the protocol for podocyte isolation (Fig. 1) were determined empirically in a series of preliminary experiments (sections 2.4.1-2.4.3, 3.1.1-3.1.3). In these experiments, the optimal concentrations of NP-suspensions for coating of podocytes on the surface of isolated glomeruli were determined, as well as the effects of different storage conditions and storage times of NPs and NP-suspensions, and the time required for the enzymatic digestion of NP-coated glomeruli.
For the further data presented in this study (sections 2.5.1.1-2.5.1.6, 3.2.1-3.2.3), podocytes were isolated from two independent cohorts of mice, following the final protocol as described below in detail. The first cohort consisted of 10, male, eight to twelve-week-old, wild-type, NMRI outbred mice. The samples generated from this cohort were used to demonstrate the appearance of isolated glomeruli and NP-coated isolated glomeruli, and to characterize the morphological and molecular properties of podocyte isolates by light- and electronmicroscopy, quantitative RT-real time PCR and Western-blot analyses (Figs. 6-12). A second cohort of mice was used to test the suitability of the described method for podocyte isolation from glomeruli of mice of different established nephropathy models, and to characterize the alterations of the negative cell surface charge of podocytes in these models (sections 2.5.2.1-2.5.2.4, 3.3). This cohort consisted of two male, 10-week-old, growth hormone (GH) transgenic NMRI outbred mice [13], two six-month-old, male transgenic CD1 outbred mice expressing a dominant negative receptor for the incretin hormone glucose dependent insulinotropicpolypeptide (GIPRdn) in pancreatic beta cells [14-16], as well as age- and sex-matched non transgenic control mice of the respective genetic backgrounds (n= 2 per nephropathy model).
2.4. Preliminary Experiments
2.4.1. Effect of Concentration Differences of NP Suspensions on the Efficiency of NP-Coating of Podocytes on the Surface of Isolated Glomeruli.
The effect of concentration differences of NP-suspensions on the NP-coating of accessible podocyte cell surfaces of isolated glomeruli was tested in a preliminary experiment. Each ~1000 glomeruli isolated from a six-month-old, male CD1 mouse were suspended in 50 µl CB and incubated with 150 µl of NP-suspensions with a concentration of 3 mg/ml, 10 mg/ml, or 30 mg/ml. The NP-suspensions were freshly prepared from vacuum-dried NPs which had been stored under oxygen-exclusion at 4-8°C for one day. Prior to incubation, the NP-suspensions were rigorously vortexed for one minute and examined under a microscope (Fig. 5). After 10 minutes of incubation with gentle agitation, the NP-coated glomeruli were filtered, washed and PAA-treated as described above and then examined by light microscopy (Fig. 7).
2.4.2. Effect of Diverse Storage Conditions on the Quality of NP-Suspensions
The effects of different storage conditions of NPs and/or NP-suspensions on their suitability for coating of podocytes on the surface of isolated glomeruli was tested in preliminary experiments, using three different NP- suspensions. All tested NP-suspensions had a concentration of 10 mg/ml CB. One NP-suspension was freshly prepared from vacuum-dried NPs (Fig. 7B), which had been stored under oxygen-exclusion at 4-8°C for six weeks. The second NP-suspension was prepared exactly as the first, but was stored in a refrigerator at 4-8°C for three weeks prior to use (Fig. 7C). The third tested NP-suspension was freshly prepared from vacuum-dried NPs, which had been stored under oxygen-exclusion at -80°C for six weeks. Prior to incubation with isolated glomeruli, the different NP-suspensions were rigorously vortexed for one minute and examined under amicroscope (Fig. 5). Then, each ~1000 glomeruli isolated from a six-month-old, male CD1 mouse were suspended in 50 µl CB and incubated with 150 µl of one of the three NP-suspensions. After 10 minutes of incubation with gentle agitation, the NP-coated glomeruli were filtered, washed and PAA-treated as described above and then examined by light microscopy. For comparison, the microscopic appearances of a NP-suspension (~10 mg/ml CB) stored for two years at 4-8°C, and of a NP-suspensions prepared from NPs stored in 50% ethanol for two years at -20°C was additionally evaluated (Fig. 7).
2.4.3. Effect of Different Time Lengths of Enzymatic Digestion of NP-Coated Isolated Glomeruli
The effect of different time lengths of enzymatic digestion of NP-coated isolated glomeruli with the trypsin- heparin-DNase I digestion solution described above at 37°C was tested in preliminary experiments, using digestion times from 10 to 40 minutes.
2.5. Main Experiments
2.5.1. Podocyte Isolation from Normal Mouse Glomeruli and Characterization of Podocyte Isolates
2.5.1.1. Isolation of Glomeruli
Glomeruli were isolated, using a modified version of a method for isolation of glomeruli from murine kidneys, perfused with spherical superparamagnetic beads [4, 17]. Mice were killed by cervico-cranial dislocation. Subsequently, 200 µl DynabeadsTM M-450 Epoxy diluted in 40 ml of 38°C PBS were perfused through the left heart ventricle, using a perfusion pressure of 70 mm Hg [18]. The kidneys were removed, decapsulated and two ~1x1x1 mm samples of cortical kidney tissue were taken from each kidney for subsequent RNA or protein analyses. The remaining tissue was minced into pieces of ~1mm³, digested with 1 mg Collagenase A/ml HBSS (20 minutes/37°C) and filtered through a 100 µm cell strainer. The glomeruli were then isolated in a strong magnetic field, applying four washes with ice-cold PBS and two washes with CB. The purity of the generated glomerulus isolates was controlled by microscopic inspection. The number of isolated glomeruli was determined by counting the number of glomeruli in an aliquot of the final glomerulus isolate. From the glomerulus isolates of ten mice, aliquots of approximately each 1000 glomeruli were put aside for mRNA and/or protein analyses and stored at -80°C until further examination. Additional sub-samples of isolated glomeruli were processed for ultrastructural examinations and immune-fluorescence analysis, as described below.

Examples for synthesis process related quality differences of silica-coated ferromagnetic nanoparticles (NPs). A: Macroscopic and microscopic (inset) appearance of a homogenous, freshly prepared suspension of well dispersed NPs, suitable for coating of isolated glomeruli. The NPs were synthetized strictly following the protocol described in the Material and Methods section and suspended in coating buffer (10 mg/ml CB). B, C: Inhomogenous suspensions of NPs with large NP-aggregates and clumps, unsuitable for podocyte isolation (compare to Fig. 7E). During the synthesis of the NPs shown in B, the ammonia solution was injected too slowly into the aqueous FeSO4 and FeCl3 solution, whereas in C, FeSO4 and FeCl3 were not completely dissolved, prior to injection of the ammonia solution. Bars = 1 cm. Bars in insets = 50 μm.
2.5.1.2. Podocyte Isolation
To specifically mark podocytes residing on the glomerular basement membrane of peripheral glomerular capillary loops, approximately 10,000-12,000 freshly isolated glomeruli per animal were diluted in 500 µl CB at 4°C, and added dropwise into 1.5 ml of the amber-coloured supernatant of a 10% (w/v) suspension of silica-coated magnetic NPs in CB taken after 15 minutes of sedimentation at 4°C. The glomeruli-NP suspension was then incubated for 10 minutes at RT, with gentle agitation, to enable the binding of the NPs to the negatively charged surfaces of the accessible podocytes upon direct physical contact. To remove unbound magnetic NPs, the mixture was then filtered through a 40 µm cell strainer and gently washed with ~5 ml of 4°C CB. Washed NP- coated glomeruli were then suspended in 500 µl 4°C CB and added dropwise into 1.5 ml of 10mg PAA/ml CB for one minute at RT to crosslink the podocyte bound magnetic NPs and neutralize their cationic surfaces. After magnetic concentration, an aliquot of the suchlike treated glomeruli was visually examined under a microscope, and counted. Another aliquot of NP-coated glomeruli was put aside for ultrastructural analyses. For enzymatic dissociation of glomerular cells, the NP coated glomeruli were then transferred into 2 ml of trypsin-heparin- DNase I digestion solution and incubated at 37°C for 25 minutes with gentle agitation. Then, the digest was carefully transferred into a Dounce tissue grinder, and trypsin activity was inactivated with 1 ml soybean trypsin inhibitor [2 mg/ml]. After careful dissociation of incompletely digested glomeruli by gentle pressure with the pestle (no strokes), the cell suspension was gently passed through a monofilament mesh with 25 µm mesh width, if appropriate. An aliquot of the cell suspension was examined microscopically to confirm the dissociation of glomerular cells/cell clusters and the NP coating of podocytes in the cell suspension. NP coated podocytes were then isolated from the cell suspension by magnetic separation, applying 1-3 careful washes with CB at 4°C. After microscopic examination and counting of cells in an aliquot of the podocyte isolate under a microscope, additional aliquots of the podocyte isolate were taken for ultrastructural examinations and immune-fluorescence analyses, as described below. For mRNA and protein analyses by real-time PCR and Western-blotting, podocyte isolates were either concentrated by magnetic isolation, or centrifuged at 800 g for 1 minute at 4°C, and the isolates, respectively cell pellets were stored at -80°C until further investigation.
2.5.1.3. Electron-Microscopy
Samples of native isolated glomeruli, of NP-coated isolated glomeruli, and of isolated podocytes retained during the different stages of the isolation procedure, were fixed in buffered 2.5% glutaraldehyde solution for >6 hours at 8°C and routinely processed for Scanning- and Transmission-Electron-Microscopy (SEM, TEM), carried out on a DSM 950 Digital Scanning Microscope, or a Zeiss EM 10 electron microscope (Zeiss, Germany), respectively. For SEM-analysis, specimen of fixed native isolated glomeruli, NP-coated isolated glomeruli, or isolated podocytes were carefully placed on a monofilament mesh with 25 µm, or 1 µm mesh width, and remained there during all following processing steps. For TEM-analysis, samples of isolated glomeruli, NP- coated glomeruli and isolated podocytes were concentrated by magnetic separation, or by centrifugation at 500 g for one minute at 4°C, prior to embedding in Epon. For TEM-analysis of APTES-modified NPs, dried NP powder was directly mixed in Epon. The average NP diameter was determined in print-outs of transmission electron micrographs and appropriate size rulers, taken at 40,000 x magnification. The diameters of 100 NPs were measured, using the VideoplanTM image analysis system (Zeiss-Kontron, Germany).
2.5.1.4. Western-Blot Analysis
Western-blot analyses of podocyte, and endothelial specific glomerular protein expression abundances were performed on samples of cortical kidney tissue, of isolated glomeruli, and of isolated podocytes from six mice. For protein extraction, tissue/cells were homogenized in PEB, heated to 95°C for 5 minutes and cooled on ice for 5 minutes. After centrifugation (18,000 g, 5 min, 4 °C), protein contents were quantified using the bicinchoninic acid method [19]. On average, 6.5 ± 2.5 µg of protein were isolated from the podocyte isolates per case. For Western-blot analyses, samples of each two mice were pooled. 10 µg of denaturated protein from each of the three pooled samples of cortical renal, glomerular, and podocyte protein, and a molecular weight standard were loaded per lane on 12% sodium dodecyl sulfate-polyacrylamide gels and separated by electrophoresis (SDS–PAGE). After electrophoresis, separated proteins were blotted to a PVDF membrane. The membrane was then cut into an upper part containing proteins with a molecular weight of more than 100 kDa, and a lower part with the proteins of less than 100 kDa, respectively. After blocking, the upper membrane part was incubated with nephrin specific antibodies, and the lower part was incubated with anti-GAPDH antibodies. After washing of the membranes, incubation with horseradish-peroxidase coupled secondary antibodies, and final washing steps, bound antibodies were visualized using ECL reagent. The upper and lower membrane parts were then stripped in SB for 40 minutes at 70°C, and re-used for detection of WT1, and CD31, respectively.
2.5.1.5. Quantitative RT-Real Time PCR (RT-qPCR) Analyses
RT-qPCR analyses were performed on samples of cortical kidney tissue, of isolated glomeruli (each~1000), and of isolated podocytes from four mice. Total RNA was isolated from the samples, using the RNeasy-Mini kit, including an on-column DNase digestion. RNA concentrations were measured on a NanoDrop 1000Spectrophotometer (Thermo Fisher Scientific, Germany). On average, 1945 ± 437 ng total RNA were isolated from the podocyte isolates per mouse. Each 500 ng of total RNA were reverse transcribed, and analyzed on an Applied Biosystems StepOnePlus Real-Time PCR System (Applied Biosystems, Germany), using Power SYBR Green PCR Master Mix, as described earlier [16]. Exon-specific PCR-primers were used for amplification of nephrin [20] (accession N°: NM_019459.2, forward primer: 5’-TCT TCA AAT GCA CAG CCA CCA-3’; reverse primer: 5’-AAA GCC AGG TTT CCA CTC CAG TC-3’), serine/cysteine peptidase inhibitor clade B member 7 (megsin, Serpinb7) [21] (forward primer: 5’-CTT CAC TGC CCT GAC CCT AAT C-3’; reverse primer: 5’-TGC AGT GCC TTG TCA ATC TGA-3’, accession N°: NM_027548.3), cd31 (pecam1) (forward primer: 5’-CTG GGA GGT CGT CCA TGT-3’; reverse primer: 5’-CAC AGG ACT CTC GCA ATC C-3’, accession N°: NM_001032378.1), wt1 [22] (forward primer: 5’-ATC TGA AGA CCC ACA CCA GG-3’; reverse primer: 5’-GCT GAA GGG CTT TTC ACT TG-3’, accession N°: NM_144783.2), and gapdh [23] (forward primer e: 5’-TGT GTC CGT CGT GGA TCT GA-3’; reverse primer: 5’-CCT GCT TCA CCA CCT TCT TGA T-3’, accession N°: NM_001289726.1). Primer sequences were queried by NCBI Blast software and the comparability of amplification efficiencies of the single primers was confirmed by performance of RT-qPCR-based standard curve analyses. All RT-qPCR measurements were performed in duplicates and included no template controls. Transcript abundances were calculated in relation to the expression Gapdh, using the 2ΔCT method [3].
2.5.1.6. Immunofluorescence-Cytochemistry and Histochemistry
For evaluation of the podocyte isolation procedure specificity and purity of the isolates, 200 µl of single cell suspensions from total glomeruli after enzymatic and mechanical cell dissociation, as well as specimen of podocyte isolates were taken from 4 mice and diluted 1:1 in 10% fetal bovine serum. Specimen for immunocyto- chemistry were prepared by cyto-centrifugation at 60 g for 3 min. The slides were air dried, fixed in 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100. Immunofluorescence-cytochemistry was performed, using specific primary antibodies for detection of nephrin and CD31, and isotype-specific secondary antibodies coupled to Cy-5, or to FITC, respectively. For positive controls, immunofluorescence-histochemistry was performed on 8 µm thick frozen sections of cortical renal tissue using the same antibodies. Embedding of slides was done with Vectashield antifade solution containing DAPI as a nuclear counterstain. The numerical fractions of cells with positive nephrin-staining in the podocyte isolates were determined by counting of 119 ± 30 cells per case in randomly selected fields of view.
2.5.2. Podocyte Isolation from Murine Nephropathy Models
2.5.2.1. Determination of Comparable Grades of Functional and Morphological Glomerular Alterations in GH-transgenic Mice and GIPRdn-Transgenic Mice
The present study examined 10-week-old GH-transgenic mice and six-month-old GIPRdn-transgenic mice in a comparable stage of morphological and functional glomerular alteration, defined by presence of albuminuria and glomerular hypertrophy [4]. Albuminuria was detected by SDS-PAGE based protein analysis of spot urine samples, and glomerular hypertrophy was confirmed by quantitative stereological determination of the mean glomerular volume, as principally described earlier [4, 16, 24]. At the examined ages, the mean glomerular volumes of GH-transgenic mice and of GIPRdn-transgenic mice were 61 ± 5%, respectively 37 ± 3% higher than in control mice.

Examples for concentration- and storage related quality differences of nanoparticle (NP)-suspensions. Aa-c: Microscopic images of different concentrations of NP in coating buffer (CB)-suspensions prepared from vacuum-dried NPs. The suspensions were stored in a refrigerator for three weeks at approximately 8°C and briefly vortexed prior to microscopic examination. Aa: Homogenous, well dispersed NP-CB-suspension (10 mg/ml), suitable for coating of isolated glomeruli (compare to Fig. 7B). Ab: NP-CB-suspension at a concentration of 3 mg/ml (too low for effective coating of isolated glomeruli, compare to Fig. 7A). Ac: Formation of undesirable NP-aggregates in a higher concentrated (30 mg/ml) NP-CB-suspension (compare to Fig. (7D, B, C): Reduced quality of NP-suspensions after prolonged storage periods. B: NP-aggregates in a NP-CB-suspension (~10 mg/ml) prepared from NPs stored in 50% ethanol for two years at -20°C. C: NP-aggregates in a NP-CB-suspension (~10 mg/ml) stored for two years at 8°C. Bars = 50 μm.
2.5.2.2. Detection of Alterations of the Negative Cell Surface Charge of Podocytes and of the Immunohistochemical Staining Intensities of Podocalyxin (PODXL) in Glomeruli of GH-Transgenic Mice and GIPRdn-Transgenic Mice
For electron-microscopic demonstration of differences of the negative cell surface charge of podocytes between GH- or GIPRdn-transgenic mice and control mice, the cationic probe polyethyleneimine (PEI) was used [25]. One GH-transgenic mouse, one GIPRdn-transgenic mouse and each one age- and sex matched control mouse of the corresponding genetic background were killed by cervico-cranial dislocation and immediately perfused with 38°C warm PBS through the left heart ventricle for one minute. The left kidney artery, vein, and ureter were then ligated and the left kidney was removed. Perfusion was continued with 4% neutrally buffered formaldehyde solution for five minutes. The right kidney was removed and post-fixed by immersion for 24 hours in the same fixation solution. Tissue samples were taken from six different locations of the cortex of the left (unfixed) kidney and further processed for PEI-labelling and transmission electron microscopy, as described earlier [25]. In each case, the capillary loops of ≥ 6 glomeruli were examined at 20-25.000 x magnification. The distribution-patterns and the glomerular abundances of PODXL, a main contributor to the glomerular polyanion, were examined by immunohistochemistry using perfusion-fixed kidney tissue samples of each one GH-transgenic mouse, one GIPRdn-transgenic mouse and of the corresponding control mice. The samples were embedded together in one single paraffin block, sectioned, and subjected to immunohistochemistry using a horseradish peroxidase-coupled secondary antibody. Slides incubated with buffer instead of the primary antibody served as negative controls.
2.5.2.3. Podocyte Isolation
Following the protocol described above (2.5.1.1-2.5.1.2), podocytes were isolated from one male, 10-week-old GH-transgenic mouse, one male, six-month-old GIPRdn-transgenic mouse, and each one age- and sex matched control mouse of the corresponding genetic background. In each case, one transversal section of the middle of one kidney was taken after perfusion with DynabeadsTM, and formalin-fixed for subsequent PODXL immunohistochemistry, performed as described above. Aliquots of isolated glomeruli, NP-coated isolated glomeruli and of podocyte isolates were successively taken during the podocyte isolation procedure and directly examined by light-microscopy to control and document the success of the single steps of the procedure, and to estimate the numbers of isolated glomeruli used for podocyte isolation, and the numbers of cells in the podocyte isolates.
2.5.2.4. Evaluation of the Purity of Podocyte Isolates by WT1 Immunohistochemistry
For histological examination and evaluation of the purity of podocyte isolates, samples of isolated glomeruli, NP-coated isolated glomeruli, and of centrifugation-pelleted podocyte isolates of the GH-transgenic mouse and its control were fixed in formalin and processed for paraffin histology (Fig. 14). Immunohistochemical detection of WT1 was performed on sections podocyte isolates and, as positive control, on sections of isolated glomeruli, using the standard avidin-biotin peroxidase complex method with diaminobenzidine (DAB) as chromogen and hemalaun as nuclear counterstain. Slides incubated with buffer instead of the served as negative primary antibody served as negative controls. The purities of the podocyte isolates were determined as the numerical fractions of immunohistochemically WT1-positive cell nuclei profiles present within the sections.
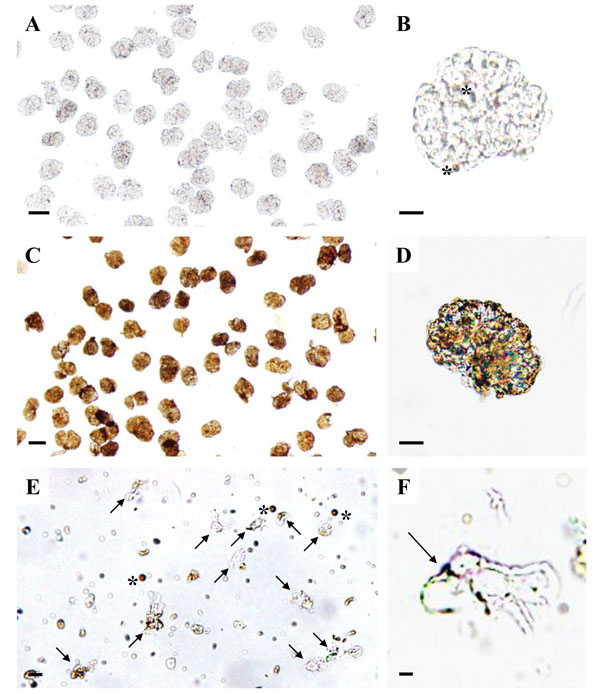
Light microscopy of podocyte isolation procedure steps. A, B: Isolated glomeruli with light-brown magnetic DynabeadsTM inside glomerular capillaries (asterisks in B). C, D: Isolated glomeruli after incubation with a 10 mg/ml suspension of cationic colloidal silica-coated ferromagnetic nanoparticles (NPs) in coating buffer and treatment with PAA. The NPs form a dense coating of the glomerular surface. E: Podocyte isolate with podocytes (arrows) and free magnetic DynabeadsTM(asterisks). F: Detail enlargement of an isolated podocyte with NP’s on the cell surface (arrow). Note the primary processes of the podocyte. Native, unstained preparations. Bars = 50 μm (A, C), 20 μm (B, D), 10 μm (E), 5 μm (F).
2.6. Data Analysis
Data were analyzed by 1-way ANOVA with Tukey‘s post hoc tests, using GraphPad Prism 5 (GraphPad Software, Inc. USA), with p<0.05 considered significant. Data are presented as means and standard errors of mean (SEM).
3. RESULTS
3.1. Preparation and Characterization of Cationic Colloidal Silica-Coated Ferromagnetic Magnetit Nanoparticles
Fig. (2) provides a schematic overview of the preparation of cationic colloidal silica-coated ferromagnetic magnetit (Fe3O4) NPs. The protocol specified in sections 2.5.1.1-2 allowed for a simple preparation of Fe3O4 NPs with strong magnetic properties within about 30 minutes. After sonication and surface cationization by modification with 3-Aminopropyltriethoxysilane (APTES), the NPs displayed stable homogeneous dispersions in coating buffer without conspicuous aggregation or subsidence for approximately 20-30 minutes (Fig. 3). In transmission electron microscopic analysis, synthesized APTES-modified NPs displayed smooth surfaces and relatively uniform diameters of 16.3 ± 3.4 nm on the average (Fig. 3).
Maintenance of oxygen free reaction conditions, the rapid injection of ammonia into the FeSO4/FeCl3 solution, and fast stirring during NP synthesis, as well as rigorous sonication of NPs prior to APTES modification appeared to be the critical steps during synthesis of cationic silica-coated magnetit NPs, essential for generation of small, well dispersed, and “sticky” NPs. Fig. (4) provides representative images of well dispersed, homogenous suspension of NPs suitable for podocyte isolation, and of NP preparations with significantly impaired qualities due to errors during NP synthesis, leading to inhomogenous suspensions with formation of larger NP-aggregates and clumps. Such NP-suspensions unsuitable for subsequent podocyte isolation (Fig. 7E) were easily identifiable by microscopic control (Fig. 4). If the critical steps of the synthesis process of NPs were observed carefully, the “quality” of NPs generated in different syntheses did not differ markedly.

Effect of different concentrations, different storage conditions, and quality differences of NP suspensions on the efficiency of NP-coating of isolated glomeruli. A: Isolated glomerulus after incubation with a 3 mg/ml NP-in-CB-suspension. Only a thin and incomplete NP-layer is present on the surface of isolated glomerulus (not suitable for subsequent podocyte isolation). B, C: Isolated glomeruli after incubation with a freshly prepared 10 mg/ml NP-in-CB-suspension (B, compare to Fig. 4A), respectively with a 10 mg/ml NP-in-CB-suspension, that had been stored at 4-8°C for three weeks (C, compare to Fig. 5Aa). All accessible outer glomerular surfaces are coated with a homogenous, dense layer of NPs (suitable for subsequent podocyte isolation). D: Isolated glomerulus after incubation with a 30 mg/ml NP-in-CB-suspension. The NP coverage of the outer surfaces of the isolated glomerulus displays an irregular thickness (not suitable for subsequent podocyte isolation). E: Isolated glomerulus after incubation with the NP-suspension shown in Fig. 4B. Multiple large NP-aggregates/clumps are attached to the glomerulus, while large areas of the glomerular surface are not covered by NPs (not suitable for subsequent podocyte isolation). Native, unstained preparations. Bars = 50 μm.
3.1.1. Effect of Concentration Differences of NP Suspensions on the Efficiency of NP-Coating of Podocytes on the Surface of Isolated Glomeruli
Incubation of isolated glomeruli with NP-suspensions of 10 mg/ml resulted in a homogenous, dense NP- coverage of all accessible outer glomerular surfaces, suitable for subsequent podocyte isolation (Fig. 7B, C). In contrast, only a thin and incomplete NP-layer was present on the surfaces of isolated glomeruli after incubation with a NP-suspension of 3 mg/ml (Fig. 7A). After incubation with a NP-suspension of 30 mg/ml, the NP coverage of the outer surfaces of isolated glomeruli was irregularly thick and formation of numerous larger aggregates of NPs not being attached to glomerular structures was observed (Fig. 7D). The adequate concentration, and distribution of NPs in the suspension could expeditiously be confirmed by microscopic examination of the NP-suspensions prior to incubation with isolated glomeruli (Figs. 4, 5).
3.1.2. Effect of Diverse Storage Conditions on the Quality of NP-Suspensions
The microscopic appearances of NP-suspensions (10 mg/ml) prepared from vacuum-dried NPs, which had been stored under oxygen-exclusion for six weeks, either at 4-8°C, or at -80°C, and of a (vortexed) NP-suspension stored at 4-8°C for three weeks (Fig. 5Aa-Ac), did not display apparent differences. The NP-coating of the outer surfaces of isolated glomeruli after incubation with these solutions was comparable, as well (Fig. 7B, C). In contrast, formation of NP-aggregates was observed in a NP-suspension (~10 mg/ml CB) stored for two years at 4-8°C, and in a NP-suspensions prepared from NPs stored in 50% ethanol for two years at -20°C (Fig. 5B, C).

Scanning electron microscopy (SEM) of podocyte isolation procedure steps. A, B: Isolated glomerulus. Podocytes are marked by arrows B: Podocyte on a peripheral glomerular capillary. Note the interdigitating foot processes of neighboured podocytes. C: Isolated glomerulus after coating with cationic colloidal silica-coated ferromagnetic nanoparticles (NPs). D: Detail enlargement of C. E: Isolated podocyte with NP’s on the cell surface (arrow). Note that some NPs will have detached from the cell surface during processing of the sample for SEM. F: Detail enlargement of foot processes of isolated podocytes. Arrow marks NPs. Bars = 1 μm.
3.1.3. Effect of Different Time Lengths of Enzymatic Digestion of NP-Coated Isolated Glomeruli
Digestion of NP-coated isolated glomeruli with the trypsin-heparin-DNase I solution for less than 20 minutes was not sufficient to achieve a sufficient pre-dissociation of glomerular cells for an adequate subsequent mechanical separation. In contrast, digestion times of more than 30 minutes led to extensive detachment of the NPs from the cell surfaces of podocytes. Adequate results were obtained applying a digestion time of approximately 25 minutes.
3.2. Podocyte Isolation from Glomeruli of Healthy Mice and Characterization of Podocyte Isolates
3.2.1. Podocyte Isolation Procedure
Incubation of isolated glomeruli with well dispersed NP suspensions (10 mg/ml) led to a homogeneous binding of NPs to all physically accessible glomerular surface structures. Using glomerulus isolates of high purities with negligible contaminations of non-glomerular kidney tissues as initial sample materials, the binding of NPs to the surface of the isolated glomeruli was almost exclusively restricted to podocytes. After cross- linking of NPs and neutralization of their cationic surface properties by poly-(acrylic) acid, the NPs formed a continuous dense layer at the surface of podocytes and their processes at peripheral glomerular capillaries, but were absent from glomerular capillary lumina, and did not pass the podocyte slit membrane, or bind to the glomerular basement membrane (GBM), or to endothelial or mesangial cells (Fig. 9). The adhesion of podocyte- bound NPs was usually stable enough to withstand the subsequent enzymatic and mechanical dissociation of individual glomerular cells, and the magnetic properties of the NPs allowed for successional isolation of NP-coated podocytes in a magnetic field (Fig. 6). During the isolation procedure, the success of the single preparation steps, i.e., the purity of glomerulus isolates, the adequate coating of the glomerular surface with NPs, the adequate dissociation of glomerular cells, the yield and the purity of the final podocyte isolates, was efficiently verifiable by microscopic inspection (Fig. 6). Following the described methodological procedure, podocyte isolation was completed within less than two hours after removal of the kidneys, with temperature conditions held at ~4°C, except for the two enzymatic digestion steps performed at 37°C for ~45 minutes.
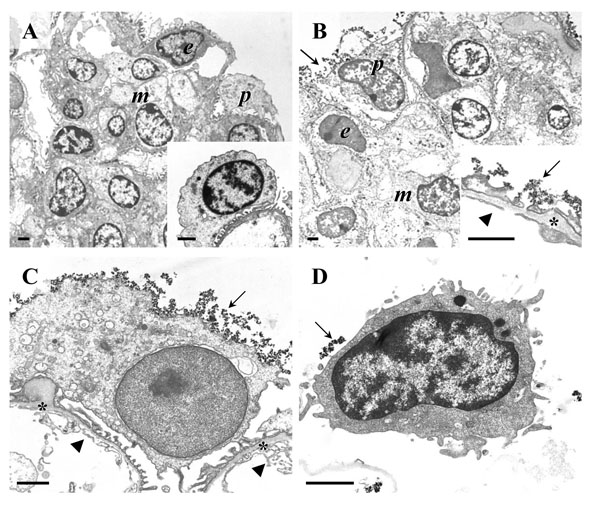
Transmission electron microscopy (TEM) of podocyte isolation procedure steps. A, B: Isolated glomeruli. Peripheral podocytes (p), endothelial cells (e), and mesangial cells (m) are indicated. A: Isolated glomerulus before coating with cationic colloidal silica-coated ferromagnetic nanoparticles (NPs). The inset to A shows a detail enlargement of a podocyte on a peripheral glomerular capillary. B: Isolated glomerulus after coating with NPs. The inset to B shows a detail enlargement of a peripheral glomerular capillary wall. C: Detail enlargement of a peripheral podocyte in a glomerulus coated with NPs. In B and C, the glomerular basement membrane (GBM) is indicated by asterisks. NPs attached to the podocyte surface, respectively the surface of podocyte foot processes are marked by arrows. Arrowheads indicate the fenestrated endothelium. Note the absence of NPs from the GBM and from endothelial and mesangial cell surfaces. D: Isolated podocyte with NP’s on the cell surface (arrow). Note that some NPs will have detached from the cell surface during processing of the sample for TEM. Bars = 1 μm.
3.2.2. Critical Steps of the Podocyte Isolation Procedure
NP-coating of isolated glomeruli, as well as the enzymatic-mechanical dissociation of NP-coated glomeruli appeared to be the two most critical steps determining the success of the method. A homogenous, well adhering, dense NP coating of podocyte surfaces without adverse agglutination of over-abundant NPs was only achieved, using solutions of well dispersed NPs in concentrations adapted to the concentration of isolated glomeruli in suspension (2.5.1.2). The effective dissociation of glomerular cells after NP-coating crucially depended on a timely terminated enzymatic digestion and careful mechanical treatment, just sufficient to separate NP-coated podocytes and free DynabeadsTM from other, non-magnetic glomerular structures. Apart from enzymatic digestion for more than 30 minutes (3.1.4), also rough mechanical dissociation, or rigorous washing steps led to detachment of NP-coatings.
3.2.3. Characterization of Podocyte Isolates
Light microscopic and ultrastructural analysis of podocyte isolates revealed single cells, cell fragments, and clusters of cells and cell fragments, identifiable as podocytes and podocyte primary processes, which were partially covered by magnetit NPs (Figs. 6, 8, 9). The podocyte preparations also contained free magnetic DynabeadsTM (Fig. 6E), which had been utilized for perfusion-embolization of glomerular capillaries for isolation of glomeruli. On average, the podocyte isolates generated from ~10,000-12,000 isolated glomeruli per mouse contained 182,000 ± 37,000 cells. Detected by immunofluorescence-cytochemistry, the majority of cells in the podocyte isolates displayed a strong immunoreactivity for the podocyte marker nephrin, whereas only few cells stained positive for the endothelial marker CD31 (Fig. 10). On average, the numerical fraction of podocytes (i.e., the fraction of nephrin-positive cells) in the isolates accounted for 63 ± 18%. Protein and mRNA analyses further confirmed the enrichment of cells with podocyte specific protein and mRNA expression properties in the podocyte isolates (Figs. 11, 12). RT-qPCR analyses showed significantly increased relative abundance levels of nephrin (Nphs1) and Wt1 mRNA in samples of isolated podocytes, as compared to samples of cortical kidney tissue. In contrast, the relative abundance levels of mRNA species specifically expressed in endothelial (Cd31, aka pecam1) or mesangial glomerular cells (megsin, aka Serpinb7) were significantly lower in podocyte isolates, than in samples of total isolated glomeruli (Fig. 12). Confirming the real-time PCR results on the protein level in Western-blot experiments, samples of isolated podocytes and samples of isolated glomeruli showed intenser WT1 and nephrin-bands, as compared to total renal cortical tissue samples (Fig. 11), whereas the intensities of the nephrin bands in samples of podocyte isolates were not obviously stronger than in samples of glomerulus isolates. In contrast, the CD31 bands displayed considerably weaker intensities in samples of isolated podocytes, as compared to protein lysates of isolated glomeruli or cortical renal tissue (Fig. 11).
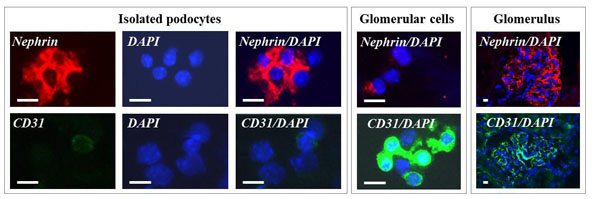
Immunocytochemistry. Detection of the podocyte marker protein nephrin, the endothelial marker protein CD31 in podocyte isolates and in total glomerular cell suspensions (glomerular cells) by immunocytochemistry and in glomeruli in frozen sections of the renal cortex (glomerulus) by immunohistochemistry. In podocyte isolates, most cells display a positive nephrin immunoreactivity (red color), while only few cells show a positive CD31 staining (green color). Nuclear stain DAPI (blue color). Bars = 5 μm.
3.3. Podocyte Isolation from Murine Nephropathy Models
As shown in Fig. (13), the examined GH-transgenic and the GIPRdn-transgenic mice displayed comparable degrees of morphological glomerular alteration, including marked glomerular hypertrophy, mesangial expansion and matrix accumulation. Immunohistochemical detection of podocalyxcin (PODXL) revealed a membranous staining pattern present in glomerular podocytes of non-transgenic control mice, as well as in GH- and GIPRdn-transgenic mice. However, in line with a potential reduction of the negative cell surface charge of podocytes, several glomerular sections of GH-transgenic and GIPRdn-transgenic mice displayed a reduced immunohistochemical PODXL staining intensity, as compared to non-transgenic control mice (Fig. 13). Electron-microscopic examination additionally revealed a slight reduction of the intensity and the density of PEI-labelled anionic sites on podocyte and podocyte foot processes surfaces in a GH-transgenic and a GIPRdn-transgenic mouse, as compared to control mice (Fig. 13). As shown for the GH-transgenic mouse and its control (Fig. 14 E-H), the binding of NPs to the outer surfaces of isolated glomeruli after incubation with the NP-suspensions was not different. Podocytes could be isolated from both the GH-transgenic, (Fig. 14) and the GIPRdn-transgenic mouse (not shown). The numbers of cells in the podocyte isolates of the GH-transgenic mouse (~185,000) and its associated control (~200,000), as well as of the GIPRdn-transgenic mouse (180,000) and the corresponding control (190,000) were in the same range.
In paraffin sections of isolated glomeruli, immunohistochemical detection of WT1 was restricted to podocytes, displaying a nuclear staining pattern (Fig. 14 M, O). The numerical fraction of cells with immunohistochemically WT1-positive nuclei in paraffin sections of podocyte isolates was 69% in the GH-transgenic mouse and 63% in the corresponding control (Fig. 14 N, P), thus lying in the same range as the purities of podocyte isolates determined by immunofluorescence based detection of nephrin-positive cells in cytological preparations of podocyte isolates of wild-type NMRI mice (3.3.2).

Western-blot detection of the podocyte marker proteins WT1 and nephrin, the endothelial marker protein CD31, and of GAPDH (loading control). Per lane, 10 μg of protein, isolated from total renal cortical tissue, from isolated glomeruli, and from isolated podocytes were loaded. Samples were pooled from each two mice (n=6). Molecular weights of protein bands are indicated. Samples of isolated glomeruli and isolated podocytes show intenser WT1 bands (arrows) and nephrin-bands, as compared to total renal cortical tissue samples. In samples of isolated podocytes, the intensities of bands referring to CD31 are weaker, as compared to samples from isolated glomeruli and total kidney cortex.
4. DISCUSSION
Our understanding of the pathogenesis of glomerular alterations leading to proteinuria and progressive loss of kidney function in a broad spectrum of renal diseases has greatly benefitted from the steadily improving level of knowledge of the mechanisms underlying the development of podocyte injury [1, 2]. Identification of relevant molecular processes governing podocyte function has long been confined by a lack of appropriate techniques for generation of sample materials allowing for a selective investigation of podocytes in certain stages of development, or disease [1, 5, 8-10]. In vitro experiments performed on podocyte cell cultures for instance, are often not adequately alienable to a respective in vivo situation, as cultured podocytes commonly display dedifferentiated morphological phenotypes with unstable variable gene expression patterns. Thus, suitable methods for direct isolation of fresh podocytes, e.g., from experimental animal models of glomerular disease, or from clinical kidney biopsy materials, are attributed a great potential to facilitate improved insights into manifold distinct aspects of podocyte cell biology and injury in vivo. In this context, methods allowing for generation of podocyte isolates for comprehensive downstream analyses of podocytic RNA- or protein expression patterns and signalling pathways are particularly interesting [5-9, 26].
So far, two principle methods for in vivo podocyte isolation extensively fulfilling these requirements have been described. Murakami et al. in (2010) used antibodies recognizing the extracellular region of nephrin to specifically label podocytes in freshly prepared mouse kidney single-cell suspensions with subsequent isolation of the antibody-bound nephrin-positive cell fraction by magnetic activated cell sorting [7]. Takemoto et al. (2006) [6] prepared single cell suspensions of isolated glomeruli from transgenic mice with podocyte specific expression of green fluorescent protein (GFP), and used fluorescence activated cell sorting for isolation of GFP expressing podocytes. The same basic principle was later applied by other investigators, who used further mouse lines expressing GFP under the transcriptional control of podocyte specific promotors to isolate podocytes for subsequent transcript profiling and proteomic analyses [8], or to directly isolate podocyte specific m-RNA by translating ribosome affinity purification (TRAP) [9].
Here, a novel method for isolation of fresh podocytes from murine kidneys for different potential downstream transcript- and protein profiling analyses is described. The isolation principle is reasonably easy: Freshly isolated kidney glomeruli are incubated with cationic silica-coated colloidal ferromagnetic NPs, which firmly bind to the accessible cell surfaces of podocytes. After dissociation of glomerular cells, NP-coated podocytes are isolated in a magnetic field. Ferromagnetic colloidal magnetit NPs with APTES modified cationized surfaces have previously been described as a useful tool for coating of cell surfaces for subsequent proteomic analysis of cell membrane proteins [27]. The presented approach for podocyte isolation is essentially different from previously published methods, since it does not employ antibodies recognizing podocyte specific surface proteins, nor does it depend on transgenic mouse lines with podocyte specific expression of fluorescent markers, such as GFP. Instead, it utilizes unique distinct anatomical and physiological properties of podocytes as differentiation criteria towards other glomerular cell types. These properties comprise (i) the localization of podocytes at the outer surface of the glomerular capillaries, where they cover the entire GBM, and (ii), their negatively charged cell surfaces. As the accessible outer glomerular surfaces in samples of intact isolated glomeruli are completely covered by podocytes, except for the vascular poles, NPs primarily get into direct contact with podocytes. This circumstance results in binding of cationic magnetic NPs to the negatively charged cell surfaces of podocytes, although the physical binding process itself is of course not podocyte-specific. Removal of excess NPs, and subsequent cross-linking of podocyte-bound NPs by poly-acrylic acid, which also neutralizes the cationic properties of their exposed surfaces, leads to formation of homogenous, dense NP- coatings of podocyte surfaces and prevents binding of NPs to other cell types during, and after subsequent enzymatic and mechanical dissociation of glomerular cells. Characterization of the final podocyte isolates proved the enrichment of cells with morphological properties specific for podocytes. These findings were additionally confirmed by RT-qPCR and Western-blot analyses, where samples of isolated podocytes displayed increased abundances of podocyte markers (vs. total cortical kidney tissue samples) and decreased abundances of endothelial and mesangial markers (vs. samples of isolated glomeruli). The finding that the intensities of the Western-blot-nephrin bands of podocyte isolates were not markedly stronger than that of samples of isolated glomeruli (Fig. 11), might likely be explained by a reduced proportion of podocyte-foot processes, where nephrin is predominantly located, and a simultaneously increased proportion of podocyte cell bodies in the podocyte isolates as compared to complete isolated glomeruli. Additionally, the exposition to NPs, chemicals and enzymes during the isolation procedure might probably also alter the antigenicity and/or, less likely, the abundance of podocyte transmembrane/cell surface proteins, such as nephrin.
Based on the underlying methodological differences, the presented NP-approach for podocyte isolation has both distinct favourable advantages, as well as specific limitations, as compared to the previously published methods of podocyte-isolation by magnetic or fluorescence activated cell sorting [6, 7]. Performance of podocyte isolation with cationic NPs is time-saving (completed in ~2 hours), and does not require large quantities of expensive antibodies, nor depend on transgenic mice with podocyte specific GFP-expression. However, compared to techniques based on isolation of antibody-tagged, or GFP expressing podocytes, the number of podocytes and the purity of the cell isolates is lower. This is partially due to the principle of the method. On the one hand, podocyte isolation with cationic NPs is principally only capable of capturing podocytes residing on peripheral glomerular capillary loops, while podocytes located in the centre of the glomerular capillary convolute are largely inaccessible. On the other hand, the method is susceptible to failure, if the critical steps of the isolation procedure do not work successfully. These steps include (i) the synthesis of well dispersed, colloidal, small, cationic, silica-coated nanoparticles, preventing agglomeration by magnetic dipole interaction; (ii) the use of appropriately concentrated NP solutions to achieve a homogenous, dense NP coating of the outer surfaces of isolated glomeruli; and, (iii), most important and most difficult to accomplish, the careful enzymatic-mechanical dissociation of NP coated isolated glomeruli, and the subsequent magnetic isolation and washing steps. While insufficient dissociation will reduce the purity of the podocyte isolates, a rough mechanical treatment after enzymatic digestion of NP-coated isolated glomeruli, as well as rigorously performed, or too often repeated washing steps will lead to detachment of the NP coating of the podocyte surface and thus to drastically reduced numbers of isolated podocytes. For reproducible results, the procedure therefore requires much practice and experience, and the effectivity of each of single steps of the isolation procedure should be controlled by microscopic examination of a small aliquot of the respective samples.
The presented approach of podocyte isolation with NPs was designed to allow for examination of RNA/protein samples generated from freshly isolated podocytes, thereby avoiding the necessity of using podocytes cultured in vitro. Therefore, the suitability of NP-isolated podocytes to be cultures in vitro was not tested in this study. However, the exposure to NPs and chemicals such as PAA during the isolation process might impair the viability of isolated podocytes, and thus limit their subsequent culturing.

RT-qPCR. Abundance of transcripts coding for the podocyte markers nephrin (NPHS1) and Wilms tumor 1 (WT1), the endothelial marker CD31, and the mesangial marker megsin (SERPINB7), relative to the abundance of Gapdh-mRNA in samples of cortical kidney tissue, isolated glomeruli, and isolated podocytes (n=4). Reflecting the marked enrichment of podocytes in podocyte isolates and the higher proportion of podocytes in samples of isolated glomeruli, as compared to total cortical kidney tissue samples, the relative Nphs1 and Wt1 mRNA abundances were the highest in podocyte isolates and the lowest in the renal cortex, with that of isolated glomeruli lying in-between. While the relative abundances of Nphs1 and Wt1 in samples of isolated podocytes were significantly higher as compared to cortical kidney tissue samples, the Nphs1 and Wt1 abundance increase in podocyte- vs. and glomerulus isolates did not reach the level of statistical significance. Corresponding to the higher proportion of endothelial and mesangial cells in the glomerular capillary tuft as compared to total cortical kidney tissue, and indicating a significant reduction of the proportion of endothelial and mesangial glomerular cells in samples of isolated podocytes, the relative Cd31 and Serpin7b mRNA abundances in samples of isolated glomeruli were significantly higher than in total cortical kidney tissue and in podocyte isolates. Although the relative Cd31 and Serpin7b mRNA abundances in samples of isolated podocytes were slightly higher than in samples of total cortical kidney tissue, these differences did not reach the level of statistical significance. Data are means ±SEM. Significant differences are indicated by asterisks. *: p<0.05; *: p<0.01; ns: not significant.
Since the method of podocyte isolation with NPs is principally based on the binding of the cationic surface of NPs to anionic sites on the cell surfaces of podocytes, a nephropathy-related reduction of the negative surface charge of podocytes could severely impede the success of the podocyte isolation method. A reduction of the negative membrane charge in the glomerular filter has been observed, e.g., in diabetic nephropathy [28], in the nephrotic stage of IgA nephropathy [29], or in the rat model of puromycin induced nephrosis [30]. Consistent with a reduced negative charge of the podocyte cell surface, the expression of the sialo- and sulfoprotein podocalyxin (PODXL), the major contributor to the anionic surface charge of podocytes [31], respectively of the sialylation of PODXL is decreased in a variety of acquired human kidney diseases [32] and experimental nephropathy models [30, 33, 34]. To examine if NPs would in principle also be suitable for isolation of podocytes from mice with pathological glomerular alterations, the method was tested using transgenic mice of two established murine nephropathy models: GH-transgenic mice, a model of progressive glomerulosclerosis [35, 36], and GIPRdn-transgenic mice, a model of diabetes mellitus associated nephropathy [16]. Transgenic mice of both models display well-characterized, progressive, glomerular alterations leading to glomerulosclerosis [16, 35, 36]. The spectrum of glomerular lesions observable in GH-transgenic and in GIPRdn- transgenic mice includes glomerular and podocyte hypertrophy, mesangial and endothelial cell hyperplasia, mesangial expansion and matrix accumulation, capillary hyalinosis and collapse, podocyte foot process effacement, albuminuria, and formation of synechiae between the glomerular capillary convolute and the capsule of Bowman. Since the glomerular lesions in GH-transgenic mice develop faster than in GIPRdn-transgenic mice, podocyte isolation was tested using a 10-week-old GH-transgenic mouse and a 6-month-old GIPRdn-transgenic mouse displaying comparable degrees of functional and morphological glomerular alteration, defined by the presence of albuminuria and glomerular hypertrophy [4]. This stage of glomerular alteration was chosen, because here the majority of glomeruli already display manifest morphologic lesions readily detectable by light microscopic examination (Fig. 13), whereas in later stages, a growing proportion of glomeruli display synechiae between the glomerular capillary convolute and the Bowman capsule. Podocytes embedded in areas of such glomerular crescents are physically inaccessible for NPs, and can therefore not be isolated by magnetic NPs, anyway.
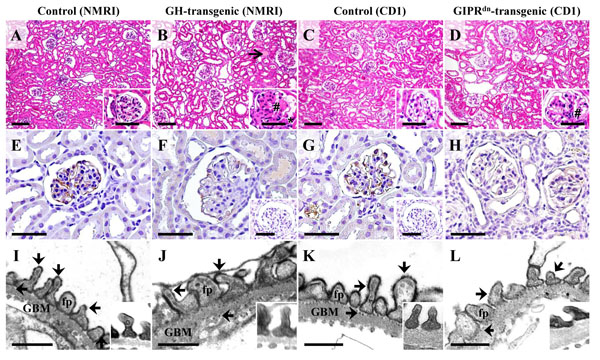
Glomerular morphology (A-D), detection of glomerular podocalyxin (PODXL) abundance by immunohistochemistry (IHC, E-H), and electron-microscopic demonstration of anionic sites on the surface of glomerular podocytes by polyethyleneimine (PEI) labeling (I-L) in a GH-transgenic mouse (male, six week-old, NMRI background, B, F, J), a GIPRdn-transgenic mouse (six-month-old, CD1 background, D, H, L), and associated age-matched control mice of the respective genetic background (A, E, I, and C, G, K). A-D: The GH-transgenic and the GIPRdn-transgenic mouse display glomerular hypertrophy and mesangial expansion and matrix accumulation (#). Additional lesions in the GH-transgenic mouse comprise glomerular capillary hyalinosis, synechiae between the glomerular capillaries and the parietal epithelium of the Bowman capsule (*), and tubular protein casts (open arrow in B). E-H: Compared to control mice (E, G), the immunohistochemical PODXL staining intensity (brown color) at the surface of glomerular podocytes is reduced in some glomeruli of the GH-transgenic mouse (F) and the GIPRdn-transgenic mouse (H). Insets to F and G: IHC negative controls. A-H: Paraffin sections, H & E staining (A-D). For IHC (E-H), DAB was used as chromogene (brown color), and hemalaun as nuclear counterstain. Bars = 50 μm, and =10 μm in insets. E, F: Transmission electron-microscopic (TEM) detection of anionic sites on podocyte surfaces with the cationic probe PEI. In non- transgenic control mice (I, K), the podocyte foot processes (fp) and the outer and inner laminae rarae of the glomerular basement membrane (GBM) display regular, punctate, electron-dense labeling patterns (arrows). In comparison, the broadened podocyte foot processes in some glomeruli of the GH-transgenic and the GIPRdn-transgenic mouse show a slightly decreased number of PEI labeled anionic sites. Insets: Podocyte foot processes of unlabeled control samples. Bars = 0.5 μm.
Detection of a reduced immunohistochemical PODXL staining intensity and reduced intensities and densities of PEI-labelled anionic sites on podocyte surfaces in some glomeruli also indicated a reduction of the anionic charge of podocytes occurring during development of glomerular alterations in GH-transgenic and GIPRdn-transgenic mice. Nevertheless, light microscopic examination of isolated glomeruli after incubation with NPs revealed no apparent differences in the binding of NPs to the surfaces of isolated glomeruli of GH-, or GIPRdn-transgenic mice and control mice. Additionally, the cells numbers in the podocyte isolates of GH-, or GIPRdn-transgenic mice were not considerably lower than in podocyte isolates of control mice. These findings show that the “adhesive” properties of the cationic surfaces of APTES-coated NPs were strong enough to bind to the surfaces of comparable numbers of podocytes in glomeruli of both examined nephropathy models. However, it has to be taken into consideration that in the examined stage of glomerular alteration, not all glomeruli, respectively, not all podocytes displayed the same degree of decreased immunohistochemical PODXL-staining intensity, or negative cell surface charge. Besides, compared to the total numbers of podocytes (est. ~2 million/mouse), the number of isolated podocytes is low (< 7%). Therefore, it cannot be excluded that individual podocytes with a greater reduction of the negative cell surface charge are indeed less efficiently isolated with cationic NPs, since this would probably not significantly affect the cell number of the final podocyte isolate. In nephropathy models with a complete, diffuse elimination of the negative cell surface charge of the majority of podocytes, other methods of podocyte isolation might therefore be more appropriate.
CONCLUSION
In summary, the described method for podocyte isolation with magnetic NPs represents a novel, fast, cost- efficient, but technically challenging approach. It is therefore regarded as an interesting alternative method for podocyte isolation, particularly in experiments, where podocyte specific antibodies or transgenic animals with podocyte specific expression of fluorescent markers are not applicable. Further experiments are considered to improve the so far relatively limited cells numbers and the low purities of the podocyte isolates, as well as studies to evaluate additional potential applications of the method, including proteomic analyses of outer podocyte cell membranes, or podocyte isolation from non-rodent species.
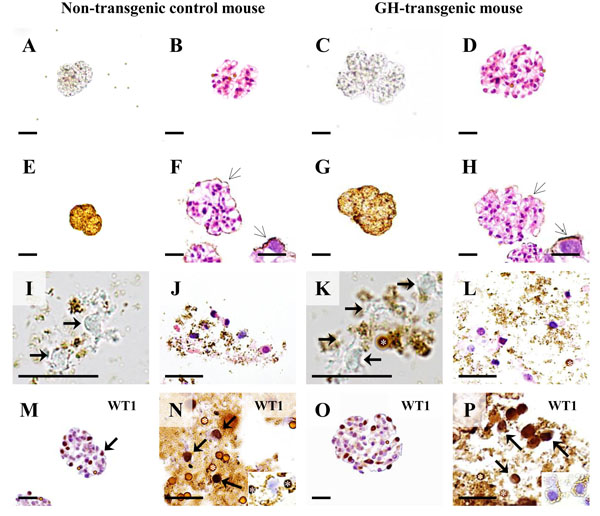
Podocyte isolation in a growth hormone (GH) transgenic mouse and non-transgenic control mouse (male, six-weeks of age). A-D: Isolated glomeruli. A, C: Native, unstained preparations. B, D: H & E stained paraffin sections. E-H: Isolated glomeruli after coating with cationic colloidal silica-coated ferromagnetic nanoparticles (NPs, open arrows). E, G: Native, unstained preparations. F, H: H & E stained paraffin sections. Arrows mark NPs on the glomerular surface. Insets: Detail enlargement of NP-covered (open arrows) podocytes. Note the larger sizes and section profiles of the glomeruli of the GH-transgenic mouse, as compared to the control mouse. I-L: Podocyte isolates. Asterisks mark DynabeadsTM. I, K: Native, unstained preparations. Arrows mark podocytes. J, L: H & E stained paraffin sections. M-P: Immunohistochemical detection of the podocyte marker protein Wilms tumor 1 (WT1) in paraffin sections of isolated glomeruli (M, O) and paraffin sections of podocyte isolates (N, P). Podocytes (arrows) display nuclear WT1-immunoreactivity. Insets to N and P: Podocyte profiles in IHC negative control sections. Asterisks mark DynabeadsTM. Bars in A-H = 50 μm, and = 10 μm in insets. Bars in I-P = 25 μm.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The author thanks Claudia Mair, Heidrun Schöl, Lisa Pichl and Adrian Ciolovan for excellent technical assistance, Dr. Robert Fux and Andrea Jahnke, (Institute of Virology) and Dr. E. Kemter and Dr. M. Dahlhoff (Institute of Molecular Animal Breeding and Biotechnology, Ludwig-Maximilians-Universität München (LMU), Germany) for providing technical equipment and antibodies, and Prof. Dr. R. Wanke (Institute of Veterinary Pathology, LMU, Germany) for review of the manuscript.

