All published articles of this journal are available on ScienceDirect.

Polypoid Change of the Glomerular Basement Membrane in a Child with Steroid Resistant Nephrotic Syndrome and ARHGAP24 Mutation: A Case Report
Authors Info & Affiliations
Abstract
Background:
Steroid resistant nephrotic syndrome (SRNS) is increasingly recognised to have a genetic basis following the identification of a number of mutations within genes encoding podocyte and basement membrane proteins. The ARHGAP24 gene product is a recently recognised important player in podocyte interaction with the glomerular basement membrane. The ARHGAP24 gene encodes a protein involved in regulating cell motility, membrane structure and polarity. Mutations in the gene have been shown in vitro to cause cell membrane ruffling.
Case Presentation:
We report a novel missense mutation in exon 4 (c.[284G>A]; p.[Arg95Gln]) of the ARHGAP24 gene in a child that presented with SRNS at four years of age. Renal biopsy demonstrated unusual polypoid changes of the glomerular basement membrane (GBM).
Conclusion:
We propose this novel ARHGAP24 mutation as causative for SRNS associated with unusual polypoid basement membrane changes. These biopsy findings, in association with ARHGAP24 mutation and clinical nephrotic syndrome are a novel finding. This finding may advance the understanding of ARHGAP24 gene product function.
BACKGROUND
Idiopathic nephrotic syndrome occurs in one to three per 100,000 children [1]. The majority of cases are steroid sensitive, but steroid resistance occurs in around 20% [1]. SRNS can be due to minimal change disease, focal segmental glomerular sclerosis or membranoproliferative glomerulonephritis as well as other rarer entities. In recent years we have realised that genetic mutations encoding proteins essential to the barrier structure and function can result in nephrotic syndrome [2]. Genetic diagnosis is important as it may predict response to therapy and is important in evaluating risk of recurrent disease following renal transplant.
This report describes unusual GBM appearances on electron microscopy seen in association with a variant in the ARHGAP24 gene. The epimembranous polypoid projections of the GBM resembled those described in five children with cyanotic congenital heart disease without nephrotic syndrome [3, 4]. The polypoid projections were seen in four of 36 autopsies of children with cyanotic congenital heart disease. The polypoid lesions increased with increasing patient age (age 1-7) and in some cases were easily visible on light microscopy. Two of the five cases had Down syndrome, however other Down syndrome children within the cohort did not display polypoid change of their GBM [4]. Both authors describe electron dense material within the polypoid projections.
The ARHGAP24 gene has been widely discussed in the literature. It is located on the long arm of chromosome 4 and has a dominant inheritance pattern. ARHGAP24 is a highly conserved gene with 3 known gene products, ARHGAP24-1 (AKA FilGAP), -2 (AKA p73RhoGAP2/RC-GAP72) and -3. The ARHGAP24 gene product is reported to affect a wide range of functions, including cell motility, membrane structure and polarity [5, 6] as well as PR interval on ECG [7], neuronal branching [8] and uterine smooth muscle contractility [9]. ARHGAP24 variants have been associated with nephrotic syndrome [10], autism [11] and disease progression in Hepatitis B virus carriers [12]. Multiple variants including at Q158R, T971, R142C, S396L, T451I and T481M, within ARHGAP24 have been found in patients with focal segmental glomerulosclerosis but not in matched controls [10]. Additionally, p.Tyr97lle has been reported as a potential cause of focal segmental glomerulosclerosis [10]. This mutation is close to the region of our patient’s variant (p.Arg95Gln).
We describe a case of SRNS in a child with a hitherto undescribed mutation in the ARHGAP24 gene, whose gene product is essential to regulating the GTPase regulating protein (GAP) involved in the glomerular filtration barrier structure and function. The ARHGAP24 variant in this child is associated with unusual polypoid change of the glomerular basement membrane.
CASE PRESENTATION
A four year old Caucasian boy presented in 2012 with nephrotic range proteinuria and oedema. His medical history was remarkable only for intermittent reactive airways disease. His urine protein:creatinine was 511 mg/mmol and a 24 hour urinary protein excretion was 1.5 grams. Creatinine was normal at <30 micromol/ L and serum albumin was 25 g/L (range 32-47) and triglycerides were high at 2.7 mmol/L (normal <1.5). Glomerulonephritis screen was negative, with anti-streptolysin O titre, anti-DNaseB titre, C3 and C4, anti-nuclear antibody and double stranded DNA antibody all within the normal range. There was no family history of note.
Oral prednisone was commenced (2mg/kg), with no remission at eight weeks. Early morning urine dipstick testing showed persistent four + proteinuria. His serum albumin at this point was 28 gm/L and creatinine remained normal. A random urine protein: creatinine ratio was 293 mg/mmol (normal <15). Clinically, the patient had ongoing fluid overload, with marked peri-orbital, scrotal and pedal oedema and intermittent tachypnoea. Steroid side effects included weight gain, hyperactivity, poor sleep and glucose intolerance.
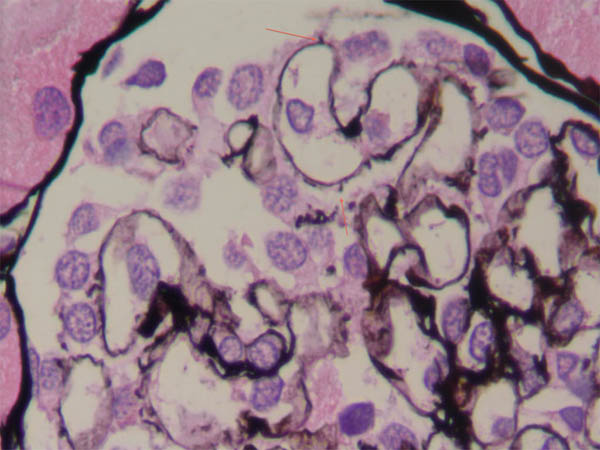
Given demonstrated steroid resistance, renal biopsy was performed. Light microscopy revealed scattered spike-like extensions of basement membrane material from the external surface of capillary loops (Fig. 1). No lesions of segmental sclerosis were identified. Immunofluorescence showed linear IgG (moderate) and lambda light chain (weak) in glomerular capillary walls, with trace mesangial IgM. There was no reactivity for IgA, C1q, C3, or kappa light chain.
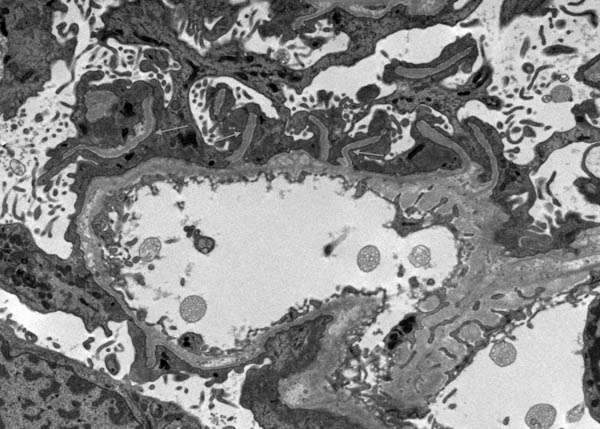
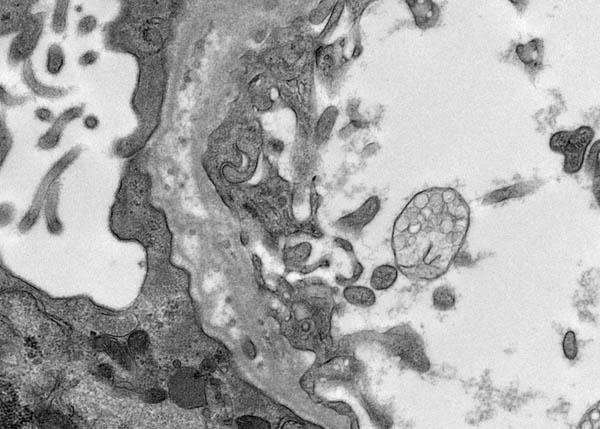
Electron microscopy showed unusual filiform projections of the glomerular basement membrane. These were fewer and longer than the spikes typically seen in membranous nephropathy (Fig. 2). There were also localised areas of glomerular basement membrane lamellation with electron-lucent areas and small electron dense granules (Fig. 3). There was extensive epithelial foot process effacement with microvillous transformation of podocytes. No electron-dense immune complex-type deposits were identified.
Oral cyclophosphamide 50mg (2 mg/kg) daily was given for 10 weeks with oral prednisone tapered over the next five months. Biochemical and clinical remission was induced by cyclophosphamide at four months from presentation with a urine protein:creatinine of 29 mg/mmol. Three years later, he remains in clinical and biochemical remission.
Screening for genetic causes of steroid resistant nephrotic syndrome was undertaken. HaloPlex target enrichment system and Next Generation Sequencing was used to test 16 genes (ACTN4, APOL1, ARHGAP24, CD2AP, COQ2, COQ6, INF2, LAMB2, LMX1B, MYO1E, NPHS1, NPHS2, PLCe1, PTPRO, TRPc6, WT1).
A heterozygous missense variant was found in exon 4 of the ARHGAP24-1 gene (c. [284G>A]; g.[86844816G>A] ; p.[(Arg95Gln)]). This is expected to cause a basic arginine residue to be substituted with a polar uncharged glutamine residue at position 95.
CONCLUSION
This is an unusual case of a child with SRNS who has polypoid projections of the glomerular basement membrane and a variant identified in the ARHGAP24 gene.
The most researched ARHGAP24 gene product, and longest protein, is called FilGAP. FilGAP includes an N terminal region with PH (pleckstrin homology), a GAP (GTPase-activating protein) activity region, a spacer, and a C-terminal coiled-coil domain [6]. The two shorter isoform gene products lack the PH region [6] where our mutation was found. This region is suspected to bind phosphatidylinositol within cell membranes and to be important in cell motility and membrane targeting [6]. This suggests the mutation affects the function of ARHGAP-1 (FilGAP) but not ARHGAP-2 and -3.
FilGAP has greatest expression in the kidney, particularly in podocytes [6, 10], which have a complex actin based cytoskeleton. FilGAP has multiple functions, including binding to FLNa (filamin A) which crosslinks actin. Actin and myosin are important in non-skeletal muscle, as they are involved in cell shape, phagocytosis, cytokinesis and cell robustness against mechanical stressors [6, 13]. FilGAP also inactivates the RAC1 protein. Unopposed activation of RAC1 results in actin-myosin interaction causing membrane protrusion, lamellipodia, cell spreading and elongated mesenchymal cell morphology [6, 10], some of which are seen in our case. Furthermore, mechanical stress has been shown in podocytes to activate the RAC1 pathway [14], which in the described mutation would be difficult to then inactivate. FilGAP also affects cell polarity [15], an important part of the glomerular filtration barrier.
The biological plausibility of this being a mutation is further aided by the characteristics of this particular ARHGAP24 missense mutation. It is a non-synonymous, rare variant in a highly conserved amino acid (up to Tetraodon) and the nucleotide is moderately conserved (phyloP: 2.71 [-14.1;6.41]). It has a minor allele frequency of 0.0002069 (dbSNP rs145434155), having been observed in 25 of 120,842 individuals across North American, European, Asian and African American populations. Further, this mutation is predicted to be pathogenic by 3 of 4 in silico models (Align GCGD: 35 (GV: 0.00 – GD: 42.81), SIFT: deleterious (score: 0, median: 4.32), MutationTaster: disease causing (p-value: 1), PolyPhen2: 0.292 (sensitivity: 0.91, specificity: 0.89). As far as we are aware, this specific mutation has not been previously reported.
In the setting of a variant in a proximate exon causing a similar phenotype, this suggests that this variant is somewhat likely to be causative. The absence of phenotypic data on other individuals with this mutation and in vitro functional validation however preclude definitive causality. A potential reliance on independently mediated intra-podocyte mechanical stress to activate RAC1 and thus mediate a disease phenotype in a two-hit pathogenic model may cause difficulty in demonstrating this phenotype in the very small numbers of individuals heterozygous for this mutation. Thus, this mutation may be viewed as conferring disease-phenotype risk within a complex genotype-phenotype relationship dependent upon the independent incidence of separate environmental factors for the emergence of a disease phenotype.
In summary, we describe a novel gene variant in ARHGAP24 whose protein product, FilGAP-1 is richly expressed in the kidney. Other mutations of this gene have been described to cause loss of polarity and cell shape, which are vital for adequate podocyte function. This has been demonstrated in vivo with foot process effacement and proteinuria. In addition, mutations in this gene have been shown to cause membrane ruffling in podocytes. It is plausible that a mutation in of c.[284G>A];[=] in exon 4 of ARHGAP24 could cause the unusual cell shape changes seen under electron microscopy in our case and the clinical presentation of steroid resistant nephrotic syndrome. Further functional assessment and in vitro mutagenesis experimentation is warranted to investigate this interesting finding.
ABBREVIATIONS
| ARHGAP24 | = Rho GTPase activating protein 24 |
| FilGAP | = Filamin A associated RhoGAP |
| FLNa | = Filamin A |
| GAP | = GTPase regulating protein |
| GBM | = Glomerular basement membrane |
| PH | = Pleckstrin homology |
| SRNS | = Steroid resistant nephrotic syndrome |
AUTHOR CONTRIBUTIONS
AF drafted the manuscript. JB conceived of the study and reviewed the manuscript. LF contributed to the manuscript, contributed the pictures and reviewed the manuscript. SM and AM contributed to and reviewed the manuscript. All authors read and approved the final manuscript.
PATIENT CONSENT
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
We gratefully acknowledge Professor John Dowling, Department of Anatomical Pathology, Alfred Hospital, Melbourne, for reviewing the biopsy.

